
Coronavirus Testing in the Outpatient Setting - CAM 380
Description
Human ccoronaviruses, first characterized in the 1960s, are named based on the spiked proteins located on their surface. As of 2020, seven coronaviruses are known to infect humans. Four, of which—229E, NL63, OC43, and HKU1—are associated with the common cold. MERS-CoV is the coronavirus that causes Middle East Respiratory Syndrome, or MERS. SARS-CoV is the causative agent of Severe Acute Respiratory Syndrome (SARS), and SARS-CoV-2 is the virus that causes coronavirus disease 2019, or COVID-19.1,2 As of June 1, 2024, the United States had reported that nearly 1.2 million people have died of COVID-19.1 Testing for a possible coronavirus infection can include molecular tests, such as nucleic acid-based testing like reverse transcription polymerase chain reaction (RT-PCR); host antibody testing; and antigen testing.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request:
- Targeted nucleic acid testing (e.g., RT-PCR, rapid molecular tests) for COVID-19 (SARS-CoV-2) is considered MEDICALLY NECESSARY in any of the following situations:
- For individuals displaying signs and symptoms of possible COVID-19 infection (See Note 1).
- For asymptomatic individuals with known exposure to COVID-19, EXCEPT when the individual has had a previous COVID-19 infection within the last 90 days.
- For individuals with signs or symptoms of SARS and who have traveled to endemic areas or who have been exposed to persons with SARS, targeted nucleic acid testing (e.g., RT-PCR) for the detection of severe acute respiratory syndrome (SARS) coronavirus RNA is considered MEDICALLY NECESSARY.
- For individuals with signs or symptoms of Middle East respiratory syndrome (MERS) and who have traveled to endemic areas or who have been exposed to persons with MERS, targeted nucleic acid testing (e.g, RT-PCR) for the detection of MERS coronavirus RNA is considered MEDICALLY NECESSARY.
- To support a diagnosis of multisystem inflammatory syndrome in children (MIS-C) (see Note 2), multisystem inflammatory syndrome in adults (MIS-A) (see Note 3), or post-acute sequelae of SARS-CoV-2 infection (PASC), nucleic acid amplification testing and host antibody serology testing is considered MEDICALLY NECESSARY.
- For symptomatic individuals, antigen-detecting diagnostic tests for SARS-CoV-2 (e.g., antigen rapid tests) once every 48 hours is considered MEDICALLY NECESSARY.
- For the diagnosis of SARS-CoV-2 reinfection, whole genome sequencing of paired specimens from distinct lineages (as defined in Nextstrain or GISAID) is considered NOT MEDICALLY NECESSARY.
- For all other situations not described above, host antibody serology testing is considered NOT MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- In the outpatient setting, SARS-CoV-2 genotyping is considered NOT MEDICALLY NECESSARY.
- For all situations, neutralization antibody testing for SARS-CoV-2 is considered NOT MEDICALLY NECESSARY.
- Testing for other endemic coronaviruses, such as 229E, NL63, OC43, and HKU1, is considered NOT MEDICALLY NECESSARY.
NOTES:
Note 1: Signs and symptoms associated with a possible COVID-19 infection can include fever, cough, fatigue, shortness of breath or difficulty breathing, congestion or runny nose, chills, muscle or body aches, headache, sore throat, new loss of taste or smell, nausea, vomiting, and diarrhea.3
Note 2: According the CDC,4 MIS-C is defined as an illness that is found in a person less than 21 years of age when all of the following conditions are met:
- Subjective or documented fever of at least 38°C;
- Clinical severity requiring hospitalization;
- Evidence of systemic inflammation indicated by elevated C-reactive protein (CRP);
- New onset of manifestations in at least two of the following categories:
- Cardiac involvement indicated by one of the following:
- Left ventricular ejection fraction < 55%.
- Coronary artery dilatation, aneurysm, or ectasia.
- Elevated troponin.
- Mucocutaneous involvement indicated by one of the following:
- Rash.
- Inflammation of the oral mucosa.
- Conjunctivitis or conjunctival injection.
- Extremity findings (e.g., erythema or edema of the hands or feet).
- Shock.
- Gastrointestinal involvement indicated by one of the following:
- Abdominal pain.
- Vomiting.
- Diarrhea.
- Hematologic involvement indicated by one of the following:
- Platelet count <150,000 cells/µL.
- Absolute lymphocyte count.
- Cardiac involvement indicated by one of the following:
Note 3: According to the CDC (CDC, 2024e), MIS-A is defined as an illness that is found in a person 21 years of age or older when all of the following conditions are met:
- Hospitalization for 24 hours or more;
- Subjective or documented fever of at least 38°C for one of the following:
- 24 or more hours prior to hospitalization.
- Within the first 3 days of hospitalization.
- No alternative diagnosis (e.g., bacterial sepsis).
- At least three of the following (occurring prior to hospitalization or within the first three days of hospitalization), with at least one being a primary clinical criterion:
- Primary clinical criteria:
- Severe cardiac illness (e.g., myocarditis, pericarditis, coronary artery dilation/aneurysm, new-onset right or left ventricular dysfunction, 2nd/3rd degree A-V block, ventricular tachycardia).
- Rash and non-purulent conjunctivitis.
- Secondary clinical criteria:
- New-onset neurologic signs and symptoms (e.g., encephalopathy in an individuals without prior cognitive impairment, seizures, meningeal signs, peripheral neuropathy including Guillain-Barré syndrome).
- Shock or hypotension not attributable to medical therapy.
- Abdominal pain, vomiting, or diarrhea.
- Thrombocytopenia.
- Primary clinical criteria:
- Evidence of SARS-CoV-2 infection;
- Evidence of systemic inflammation (elevated CRP, ferritin, interleukin-6, erythrocyte sedimentation rate, or procalcitonin).
Table of Terminology
| Term |
Definition |
| 2019-nCoV |
2019 novel coronavirus |
| AACC |
American Association for Clinical Chemistry |
| AAP |
American Academy of Pediatrics |
| ACE-2 |
Angiotensin converting enzyme-2 |
| ACR |
American College of Rheumatology |
| ACS |
American Chemical Society |
| Ag-RDTs |
Antigen-detecting rapid diagnostic tests |
| AMA |
American Medical Association |
| APSF |
Anesthesia Patient Safety Foundation |
| ARDS |
Acute respiratory distress syndrome |
| ASA |
American Society of Anesthesiologists |
| ASM |
American Society for Microbiology |
| BAL |
Bronchoalveolar lavage |
| BNP |
B-type natriuretic peptide |
| CARES Act |
Coronavirus Aid, Relief, And Economic Security Act |
| Cas12a |
CRISPR associated protein 12a |
| CBC |
Complete blood cell count |
| CDC |
Centers For Disease Control and Prevention |
| cDNA |
Complementary deoxyribonucleic acid |
| CFR |
Code of Federal Regulations |
| CI |
Confidence interval |
| CLIA |
Chemiluminescence enzyme immunoassay |
| CLIA ’88 |
Clinical Laboratory Improvement Amendments Of 1988 |
| CMS |
Centers For Medicare & Medicaid Services |
| COVID-19 |
Coronavirus disease 2019 |
| CPK |
Creatine phosphokinase |
| CRP |
C-reactive protein |
| CSSE |
Center for Systems Science and Engineering |
| CT |
Cycle threshold |
| cVNT |
Competitive neutralization test |
| DNA |
Deoxyribonucleic acid |
| DPP7 |
Dipeptidyl peptidase 7 |
| ECDC |
European Centre for Disease Prevention and Control |
| ESR |
Erythrocyte sedimentation rate |
| ETS |
Emergency temporary standard |
| EU/EEA |
European Union / European Economic Area |
| EUA |
Emergency use authorization |
| FAQ |
Frequently asked questions |
| FDA |
Food and Drug Administration |
| FET |
Field-effect transistor |
| FIA |
Fluorescence immunoassays |
| Flu SC2 |
Influenza SARS-CoV-2 (multiplex assay) |
| FN |
False negative |
| FP |
False positive |
| GISAID |
Global initiative on sharing all influenza data |
| GOLGA3 |
Golgi autoantigen, golgin subfamily a, 3 |
| GRADE |
Grading Of Recommendations Assessment, Development, and Evaluation |
| HCoV |
Human coronavirus |
| HCP |
Health care personnel |
| HCW |
Healthcare worker |
| HHS |
Health And Human Services |
| HKU1 |
Human coronavirus |
| HLA |
Human leukocyte antigen |
| HSCT |
Hematopoietic stem cell transplant |
| ICMA |
Immunochemiluminometric assay |
| ICR |
Investigative criteria for suspected cases of SARS-CoV-2 reinfection |
| IDSA |
Infectious Diseases Society of America |
| IFU |
Instructions for use |
| IgA |
Immunoglobulin A |
| IgG |
Immunoglobulin G |
| IgM |
Immunoglobulin M |
| IL-1 |
Interleukin 1 |
| IL-6 |
Interleukin 6 |
| INR |
International normalized ratio |
| IQR |
Interquartile range |
| IVIG |
Intravenous immunoglobulin |
| JAMA |
Journal of the American Medical Association |
| LDH |
Lactic acid dehydrogenase |
| LDTs |
Laboratory-developed tests |
| LFIAs |
Lateral flow immunoassays |
| LoD |
Limit of detection |
| MERS |
Middle east respiratory syndrome |
| MERS-CoV |
Middle east respiratory syndrome–related coronavirus |
| MHRA |
Medicines & Healthcare Products Regulatory Agency |
| MIS-A |
Multisystem inflammatory syndrome in adults |
| MIS-C |
Multisystem inflammatory syndrome in children |
| MMWR |
Morbidity And Mortality Weekly Report |
| MT |
Mid-turbinate |
| N |
Nucleocapsid |
| NAAT |
Nucleic acid amplification test |
| NAb |
Neutralizing antibody |
| NGS |
Next-generation sequencing |
| NIH |
National Institutes of Health |
| NP |
Nasopharyngeal |
| NPA |
Negative percent agreement |
| NT-proBNP |
N-terminal pro hormone BNP |
| NW |
Nasopharyngeal wash/aspirate or nasal wash/aspirate |
| OD |
Optical density |
| OP |
Oropharyngeal |
| opvCRISPR |
One-pot visual SARS-CoV-2 detection system |
| OSHA |
Occupational Safety and Health Administration |
| PASC |
Post-Acute Sequelae Of SARS-CoV-2 Infection |
| PCR |
Polymerase chain reaction |
| PEM |
Post-exertional malaise |
| PHE |
Public Health England |
| PHS Act |
Public Health Service Act |
| POC |
Point-of-care |
| POC/NP |
Point of care/near person |
| PPA |
Positive percent agreement |
| PPE |
Personal protective equipment |
| pro-BNP |
Pro hormone B-type natriuretic peptide |
| PSO |
Past symptom onset |
| PT |
Prothrombin time |
| PTT |
Partial thromboplastin time |
| ptxP |
Single-copy promoter target |
| RADT |
Rapid antigen detection test |
| RBD |
Receptor binding domain |
| RdRp |
Ribonucleic acid-dependent ribonucleic acid polymerase |
| RNA |
Ribonucleic acid |
| RP |
Ribonuclease P gene |
| RP |
Respiratory pathogen |
| RP2 |
Respiratory panel 2 |
| RP2.1 |
Respiratory panel 2.1 |
| RT |
Reverse transcriptase |
| RT-LAMP |
Reverse transcription loop-mediated isothermal amplification |
| RT-PCR |
Reverse transcription polymerase chain reaction |
| SARS-CoV |
Severe acute respiratory syndrome- coronavirus |
| SARS-CoV-2 |
Severe acute respiratory syndrome coronavirus 2 |
| SHEA |
Society for Healthcare Epidemiology of America |
| SNP |
Single nucleotide polymorphism |
| SOT |
Solid organ transplant |
| ssDNA |
Single-stranded deoxyribonucleic acid |
| sVNT |
Surrogate viral neutralization test |
| TCID50 |
Median tissue culture infective dose |
| TMA |
Transcription-mediated amplification |
| TMEM189–UBE2V1 |
PEDS1-UBE2V1 readthrough |
| TN |
True negative |
| TP |
True positive |
| UCSD |
University of California San Diego |
| VOC |
Variant of concern |
| WGS |
Whole genome sequencing |
| WHO |
World Health Organization |
Reimbursement
- AMA standard practice for COVID-19 testing states not to include both the HCPCS and AMA code for the same procedure on the same DOS and that only one code should be used, therefore only one code per date of service will be reimbursed.
- Specimen collection codes for coronavirus testing are considered incidental and will not be reimbursed
Rationale
On March 11, 2020, the World Health Organization (WHO) declared the novel coronavirus SARS-CoV-2, or COVID-19, a global pandemic (Cucinotta & Vanelli, 2020). COVID-19 is the third recent human coronavirus to be declared an emergency. SARS (Severe Acute Respiratory Syndrome) was recognized as an emergency by the WHO in February 2003 (WHO, 2024b). This outbreak in 2003 resulted in over 8000 cases in 26 different countries. Since 2003, only four limited reoccurrences have been reported according to the WHO — three incidences are due to laboratory accidents (in Taipei and Singapore) and one incident of undetermined source in China (WHO, 2024b). As early as September 2012, another human coronavirus, MERS-CoV, began to spread in the Middle East, causing Middle East Respiratory Syndrome (MERS). Although the WHO did not initially declare MERS an emergency, they have since added MERS to their list of pandemic/epidemic diseases. Since September 2012 and as of the end of October 2021, the WHO reports 2574 laboratory-confirmed cases of MERS with 858 MERS-associated deaths (34.4% fatality rate) in 27 countries (WHO, 2024a).
Unlike the initial SARS and MERS outbreaks that were predominantly regionally contained, COVID-19 became a global pandemic. According to the WHO, as of Sept. 27, 2023, there were more than 770 million confirmed cases of COVID-19 with over 6,959,316 confirmed deaths worldwide (WHO, 2023). Infection from the novel human coronavirus SARS-CoV-2 can result in coronavirus disease 2019 (COVID-19). The WHO reports approximately 15% of individuals with COVID-19 develop severe disease requiring oxygen support while 5% develop “critical disease” with complications such as respiratory failure or multiorgan failure (WHO, 2021b). Older individuals and patients with comorbidities — such as cardiovascular disease, diabetes mellitus, hypertension, chronic lung disease, cancer, chronic kidney disease, obesity, and smoking — have an increased likelihood of poor outcomes (Gandhi, 2024). Sepsis, multiorgan failure (including the kidney, liver, and heart), pneumonia, and acute respiratory distress syndrome (ARDS) can also occur (WHO, 2021b; Yang et al., 2020). Severe outcomes have been associated with the following laboratory features: lymphopenia, elevated liver enzymes, elevated lactate dehydrogenase (LDH), elevated inflammatory markers (such as CRP and ferritin), elevated D-dimer, elevated prothrombin time (PT), elevated troponin, elevated creatine phosphokinase (CPK), and acute kidney injury (Gandhi, 2024).
Much of what has generated this global pandemic is attributed to the different levels of transmissibility of the SARS-CoV-2 virus compared to SARS-CoV-1 and MERS, which can arise from the viral load. Simply put, viral load is the number of viral particles/virions in a milliliter of blood (Ryding, 2020). The viral load of SARS-CoV-2 “peaks around the time of symptom onset, followed by a gradual decrease to a low level after about 10 days. Regarding the period of high infectiousness, a recent study reported that exposure to an index case within five days of symptom onset confers a high risk of secondary transmission” (Kawasuji et al., 2020). This finding was corroborated by other studies, which found that “SARS-CoV-2 viral load in the upper respiratory tract appeared to peak in the first week of illness, whereas that of SARS-CoV peaked at days 10 – 14 and that of MERS-CoV peaked at days 7 – 10;” because SARS-CoV-2 viral load peaks faster, it can be more transmissible earlier in the disease course (Cevik et al., 2021). However, after reaching its peak during symptom onset, the viral load decreases “monotonically” (Kawasuji et al., 2020). If viral loads do not decrease, patients will be more likely to suffer worse outcomes and require hospitalization (Griffin, 2020). Viral load has been found to be either similar among symptomatic and asymptomatic COVID-19 positive individuals, or higher among symptomatic individuals (Kawasuji et al., 2020). Infectiousness of COVID-19 also correlates with shedding, meaning that the viral particles can replicate in an individual and spread in the environment to others. The mean duration of SARS-CoV-2 RNA shedding “was 17.0 days (95% CI 15·5 – 18·6; 43 studies, 3229 individuals) in upper respiratory tract, 14.6 days (9·3 – 20·0; seven studies, 260 individuals) in lower respiratory tract, 17.2 days (14·4 – 20·1; 13 studies, 586 individuals) in stool, and 16.6 days (3·6 – 29·7; two studies, 108 individuals) in serum samples,” with maximum shedding duration reaching “83 days in the upper respiratory tract, 59 days in the lower respiratory tract, 126 days in stools, and 60 days in serum”(Cevik et al., 2021).
In children and adolescents, reports of a multisystem inflammatory syndrome (MIS-C) with similarities to Kawasaki disease and toxic shock syndrome have been linked to COVID-19 (DeBiasi et al., 2020; Jones et al., 2020; Verdoni et al., 2020; WHO, 2020c). Multisystem inflammatory syndrome has also been reported in adults (MIS-A). From June to October 2020, researchers reported 27 cases of MIS-A in the US and UK (Baum, 2020). The case definition of MIS-A includes “(1) hospitalization without evidence of severe respiratory illness (to exclude hypoxia as the cause of the signs and symptoms), (2) extrapulmonary organ system involvement (including hypotension or shock, cardiac dysfunction, arterial or venous thromboembolism, acute liver injury, or dermatologic abnormalities), and (3) laboratory evidence of acute inflammation (e.g., highly elevated C-reactive protein, ferritin, D-dimer, or interleukin-6)” (Baum, 2020). Most patients present with a fever > 100.4 °F, cardiac abnormalities (arrhythmias, elevated troponin levels, or left or right ventricular dysfunction), and gastrointestinal symptoms. Rare symptoms include dermatological manifestations or respiratory symptoms such as pleural effusion. Patients may have elevated laboratory markers of inflammation including CRP, ferritin, and markers of coagulopathy including D-dimer (Morris et al., 2020).
As SARS-CoV-2 has continuously mutated over the course of the pandemic, CDC has adjusted their categorizations of the numerous variants based on shared attributes that may require public action and on available information. CDC lists four variant classifications on their website: variants being monitored (VBM), variants of interest (VOI), variants of concern (VOC), and variants of high consequence (VOHC). VBMs are described as “lineages with potential impact on available medical countermeasures based on analysis of genetic sequence data,” “lineages that previously caused more severe disease or increased transmission but that are no longer detected”, “lineage with an unusually large number of antigenic mutations AND presence in multiple countries with collection dates within 4 weeks”, or “lineages previously designated as a VOI, VOC, or VOHC that are currently circulating at very low levels in the United States.” As such, VBMs are “no longer circulating at sustained levels and no longer poses significant risk to public health in the United States” and VOIs and VOCs may be downgraded to this list when evidence suggests that they no longer pose significant risk to public health (CDC, 2024a). The list of possible attributes for variants of interest (VOIs) include the presence of “specific genetic markers that are predicted to affect transmission, diagnostics, therapeutics, or immune escape”, and “evidence that it is the cause of an increased proportion of cases or unique outbreak clusters.” In addition to including possible features of VOIs, VOCs are marked by a “increase in transmissibility,” “more severe disease (for example, increased hospitalizations or deaths),” “significant reduction in neutralization by antibodies generated during previous infection or vaccination,” and “reduced effectiveness of treatments or vaccines, or diagnostic detection failures.” A VOHC “has clear evidence that prevention measures or medical countermeasures (MCMs) have significantly reduced effectiveness relative to previously circulating variants” (CDC, 2024a). Currently, all the variants being monitored by CDC fall in VBM status except for the Omicron strain (B.1.1.529 and descendant lineages), which is labeled a VOC.
The CDC indicates three vaccines as authorized and recommended to prevent COVID-19 in the US: Pfizer-BioNTech COVID-19 Vaccine, Bivalent; Moderna COVID-19 Vaccine, Bivalent; and Novavax COVID-19 Vaccine, Adjuvanted. The Pfizer-BioNTech and Moderna COVID-19 vaccines are mRNA vaccines, which instruct B and T lymphocytes to fight off that specific mRNA-encoded protein from COVID-19 in the event of future exposure. Novavax is a protein subunit vaccine that delivers pieces (spike proteins) of the virus that causes COVID-19, as well as an adjuvant that helps the immune system respond in the event of future exposure (CDC, 2024c).
Besides the viruses associated with SARS, MERS, and COVID-19, four other human coronaviruses (HCoVs) are currently known — 229E, NL63, OC43, and HKU1. These four viruses are considered endemic to the human population, and they typically cause mild respiratory tract infections associated with the common cold; in fact, it is approximated that up to one-third of all “common colds” may be due to one of these four endemic human coronaviruses. These HCoVs can cause both upper and lower respiratory infections, but they typically result in relatively mild, or even asymptomatic, cases. In immunosuppressed individuals, including those with pre-existing pulmonary diseases, progression to acute respiratory failure can occur in some cases (Corman et al., 2019; Ludwig & Zarbock, 2020).
Nucleic Acid Testing for Human Coronavirus Infections
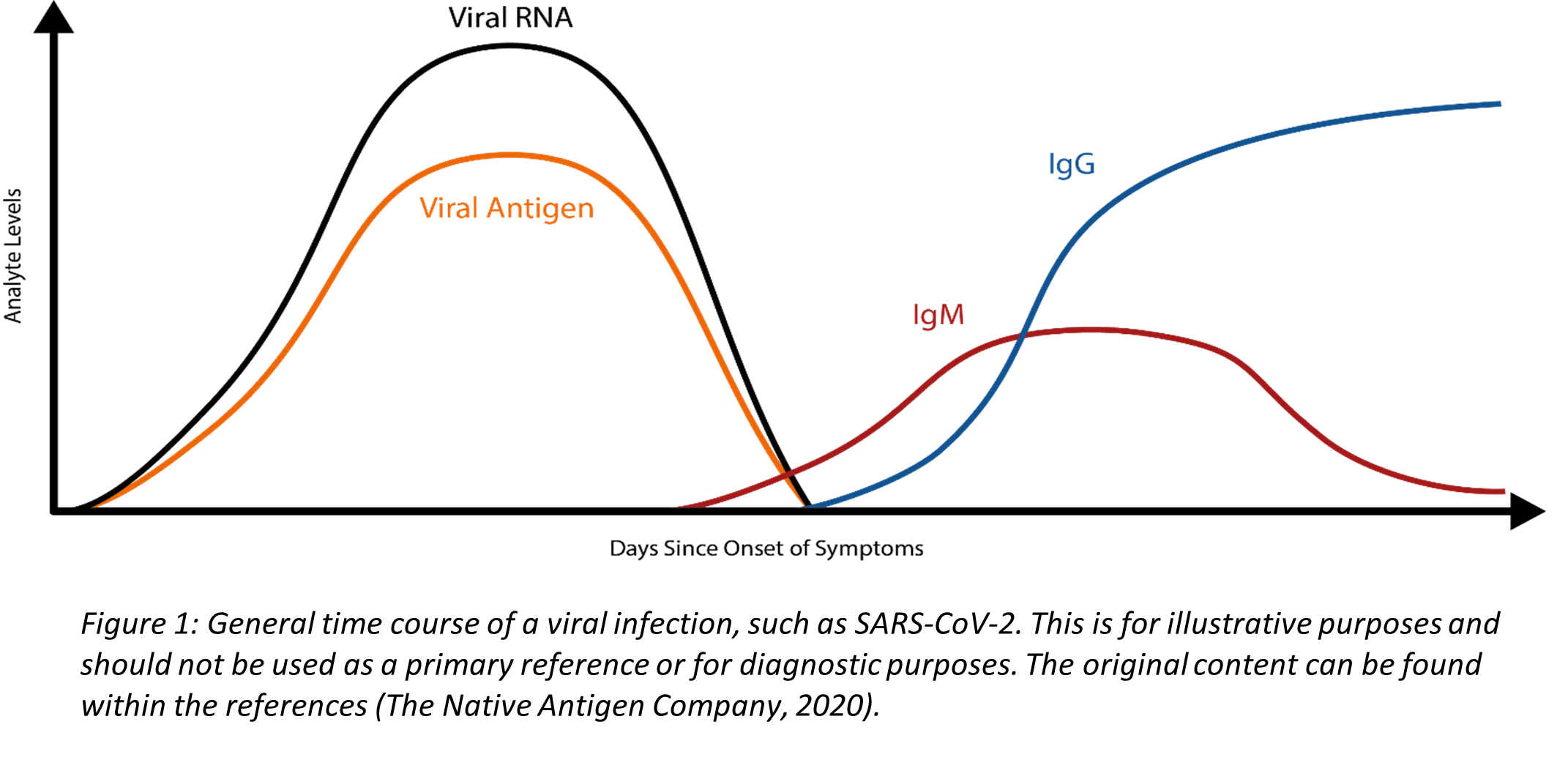
Coronaviruses are a family of enveloped, single-stranded positive-sense RNA viruses. During the initial phase of infection, the virus can be detected in respiratory specimen due to high concentrations of viral RNA (Figure 1). RT-PCR is a powerful molecular technique that synthesizes complimentary DNA (cDNA) from the initial RNA template and uses primers to manufacture multiple cDNA copies for analysis. RT-PCR, when used with appropriate primers targeting the SARS-CoV-2 RNA, is used to diagnose an acute infection. The CDC RT-PCR Diagnostic Panel detects SARS-CoV-2 virus in the upper and lower respiratory specimen. As depicted in Figure 1, the concentration of viral RNA decreases as the immune system fights the infection, and very low or undetectable viral RNA levels are typically present after an individual has recovered. Consequently, RT-PCR cannot be used to screen for a past infection. Another limitation to RT-PCR is that it does require specific instrumentation, and, therefore, is less amenable as a rapid, point-of-care test. RT-PCR results of SARS-CoV-2 may fluctuate and become unstable over time, thus requiring other clinical diagnostic measures, such as computerized tomography imaging to supplement isolation, discharge, and any transfers during this epidemic (Li et al., 2020).
Clinical Utility and Validity of Nucleic Acid Testing
Many studies have been performed to date to evaluate the analytical performance of RT-PCR. One study, using a high-throughput platform, for example, reported a limit of detection (LoD) of 689.3 copies/mL and 275.72 copies per reaction at 95% detection probability (Pfefferle et al., 2020). The WHO diagnostic RT-PCR test utilizes two genes--the E gene as the molecular target (where the limit is 3.9 copies per reaction) and the RdRp gene as the molecular target (limit of 3.6 copies per reaction) (Lippi et al., 2020). One recent study reported possible in vitro cross-reactivity between the RdRp-based method used predominantly in European labs with SARS-CoV in cell culture (Chan et al., 2020). SARS-CoV is the coronavirus that caused the initial SARS (Severe Acute Respiratory Syndrome) outbreak in 2003 (WHO, 2024b). The likelihood of either a co-infection of SARS-CoV and SARS-CoV-2 or a concurrent outbreak of both viruses is small. The CDC diagnostic panel test does not target the RdRp gene; it consists of two primer/probe sets of the N gene and one primer/probe set for human RNase P gene (RP) as the control. The CDC diagnostic panel has a reported limit of 1.0 – 3.2 copies/µL (Lippi et al., 20
Reports of initial negative RT-PCR results in individuals who later develop symptomatic COVID-19 have been published, but this may occur if the sample was not properly collected or if it was taken from the patient early in the infection during the initial incubation period of SARS-CoV-2, which is approximately six days (interquartile range [IQR], 2 – 11 days) (Backer et al., 2020; Lippi et al., 2020). Consequently, it is important to remember that “Negative results do not preclude SARS-CoV-2 infection and should not be used as the sole basis for patient management decisions. Negative results must be combined with clinical observations, patient history, and epidemiological information” (LabCorp, 2022a, 2022b).
To compare and analyze the diagnostic efficacy of two RT-PCR test kits for detection of SARS-CoV-2, Lu et al. (2020) studied throat swab samples from 18 hospitalized patients with a clinical COVID-19 diagnosis and 100 hospitalized patients without COVID-19 diagnosis. Two different RT-PCR tests from Sansure Biotech Inc (SansureBiotech, 2022) and Shanghai BioGerm Biotechnology Co., Ltd (BioGerm, 2024) were used. Table 2 (Lu et al., 2020) shows that the detection efficacy of the BioGerm PCR kit was higher than that of the Sansure PCR kit. These two kits had the same specificity and positive predictive value, but the sensitivity of the Sansure PCR kit was 83.3%, whereas the sensitivity of the BioGerm PCR kit was 94.4%. For the Sansure PCR kit, three of the 18 samples were false-negative results, and for the BioGerm PCR kit, one of the 18 samples was a false-negative result. No false-positive results were detected in these tests. The author suggests that “these findings provide important information for the ongoing optimization of viral detection assays following the emergence of COVID-19” (Lu et al., 2020).
| COVID‐19 samples (n = 18) |
None‐ COVID‐19 samples (n = 100) |
Sensitivity (95%CI) |
Specificity (95%CI) |
PPV (95%CI) |
NPV (95%CI) |
Kappa (95%CI) |
|||
| Test kits |
Positive |
Negative |
Positive |
Negative |
|||||
| Sansure |
15 |
3 |
0 |
100 |
0.833(0.577‐0.956) |
1.000(0.954 – 1.000) |
1.000(0.747‐1.000) |
0.971(0.911 – 0.992) |
0.894(0.726 – 1.000) |
| BioGerm |
17 |
1 |
0 |
100 |
0.944(0.706‐0.997) |
1.000(0.954 – 1.000) |
1.000(0.771‐1.000) |
0.990(0.938 – 0.999) |
0.966(0.880 – 1.000) |
Table 2. Diagnosis efficacy of Sansure and BioGerm test kits for SARS‐CoV‐2 nucleic acid detection.
In a case series study of multisystem inflammatory syndrome in adults (MIS-A) associated with SARS-CoV-2 infection, 16 patients ranging from 21 to 50 years old were enrolled and tested with PCR assay. Ten out of 16 patients had positive SARS-CoV-2 PCR test results at the time of admission. Two patients had positive SARS-CoV-2 PCR test results 14 and 37 days before admission and negative PCR results at the time of admission. Three patients had positive SARS-CoV-2 PCR test results 25 – 41 days before admission and continued positive PCR test results at the time of admission. “Given the high proportion of MIS-C patients with negative PCR testing, clinical guidelines recommend the use of both antibody and viral testing to assist with diagnosis” (Morris et al., 2020).
Li et al. (2021) conducted a cross-sectional analysis on 30 patients with COVID-19 diagnoses to compare the sensitivity of SARS-CoV-2 testing in anterior nasal vestibular swabs versus oropharyngeal swabs. After specimen collection, RT-PCR assays were used to test them for SARS-CoV-2. They found that 56.7% of the patients tested positive using oropharyngeal specimen, whereas 66.7% of patients tested positive with the nasal swab specimens. Ultimately, there is “adequate sensitivity” to use the less invasive anterior nasal vestibular swabs to detect COVID-19 infection confirmed by RT-PCR (Li et al., 2021).
Yau et al. (2021) evaluated the clinical utility of a rapid “on-demand” PCR-based testing service in an acute hospital setting. To increase hospital efficiency starting from July 2020, the researchers focused on moving patients quickly to isolation rooms and minimize potential risk of transmission in crowded areas. From their study, it was found that the “daily/monthly PCR positive test numbers approximately followed the local and national UK trend in COVID-19 case numbers, with the daily case numbers being reflective of the Nov and Dec 2020 surges.” It ultimately helped to reduce “unnecessary ‘length-of-stay’ in a busy acute respiratory ward.” Patients were able to be rapidly separated based on COVID-19 positive diagnosis and the system in place reduced exposure and nosocomial transmission (Yau et al., 2021).
Dighe et al. (2022) studied a lateral flow strip-based RNA extraction and amplification-free nucleic acid test (NAT) for rapid diagnosis of COVID-19 at point of care which takes no longer than 30 minutes. This test uses highly specific 6-carboxyfluorescein (6-FAM) and biotin labeled antisense oligonucleotides (ASOs) as probes those are designed to target the N-gene sequence of COVID-19. This study evaluated 60 samples using the lateral flow assay and results were compared with the FDA approved TaqPath RT-PCR kit. According to the results, the assay obtained almost 99.99% accuracy and specificity. The authors conclude that this new LFA method could be "expanded beyond COVID-19 detection, simply by altering its targeting antisense oligonucleotides, to become a global health technology that contributes to providing low-cost diagnostics" (Dighe et al., 2022).
Mawhorter et al. (2022) investigated the impact and cost of a routine pre-operative COVID-19 PCR testing algorithm for asymptomatic patients before elective surgery at a rural academic institution per recommendations by the American College of Surgeons. From 7,579 pre-procedural tests that were completed since May 2020 using the protocol, the study yielded 31 (0.41%) positive results in asymptomatic patients. With these positive results, there were impacts on both the cost and delay of the procedure. The results showed that “20 procedures (62.5%) were delayed an average of 49 days, 8 were not performed, and 3 proceeded without delay,” with a prolonged delay for the three urological procedures of 59 days. They also identified that the number needed to test for one positive result was 244, with $11,573 as cost for each positive result. This analysis found that the hospital was able to be more cost-effective (each test was $34 – $54) with a standardized testing algorithm prior to procedure performance (Mawhorter et al., 2022).
Host Antibody Testing
The COVID-19 illness begins with an initial infection by SARS-CoV-2. Viral invasion stimulates the host immune response to produce immunoglobulins, such as IgM, IgA, and IgG, that can target the invading virus. However, there is a delay between the time of initial infection and the production of immunoglobulins (Figure 1) (The Native Antigen Company, 2020). Typically, several days after the initial onset of symptoms, the first IgM immunoglobulins are produced to combat the viral infection. IgA (not shown in Figure 1), immunoglobulins secreted to protect predominantly the mucosal linings of the gastrointestinal, respiratory, and genitourinary tracts (Woof & Kerr, 2006), typically have a half-life of four to six days (Morell et al., 1973). Finally, IgG, the long-term immunoglobulins found within body fluids that fight bacterial and viral infections, are produced and IgM production wanes. Some limited studies have indicated that some individuals may initially produce IgM and IgG antibodies concurrently, but additional research is needed (Padoan et al., 2020).
Serological host antibody tests can detect the presence of IgM and IgG antibodies that an individual has developed in response to an infection — in this case, a SARS-CoV-2 viral infection. The test may report total antibodies present, meaning either it does not distinguish between IgG and IgM or that it is reporting the sum of IgG and IgM. This is sometimes referred to as “total antibody testing.” On the other hand, the test may be specific for one antibody, such as IgG or IgM, or the test may claim to accurately distinguish between the antibodies.
Another type of antibody testing is “neutralizing” antibody detection, as opposed to “binding” antibody detection described above. This process involves incubating serum with a live version of the virus. The analytes of interest are the antibodies that have the capability to prevent infection by the virus (i.e., neutralization). Identification of these antibodies may contain useful clinical information and are often reported in an aggregate titer, as opposed to specifying each individual antibody (Espejo et al., 2020).
Clinical Utility and Validity of Host Antibody Testing
Antibody testing has many potential uses. Ideally, the use of an accurate, reliable antibody test could possibly show whether someone has previously been exposed to the virus. This could indicate possible immunity in an individual. Please note that the antibody test is not used as a diagnostic test, meaning it should not be used to diagnose an acute infection. Within the FDA policy for diagnostic testing for COVID-19, issued on Nov. 15, 2021 they state, “Results from antibody testing should not be used to diagnose or exclude SARS-CoV-2 infection” (FDA, 2023b).
Since SARS-CoV-2 is a new, emerging virus, it is not known for certain how long it takes for the seroconversion to occur or when antibodies start to appear in the blood at high enough concentrations for accurate testing results. A recent study published in Clinical Infectious Diseases reports an average of seroconversion time for IgM and IgG at 12 and 14 days, respectively (Zhao et al., 2020). A small study (n = 34 patients) reports the presence of IgG for at least seven weeks (the duration of the study) (Xiao et al., 2020). Another study, however, reports that IgM testing has similar, if not better positive detection rate than PCR 5.5 days after initial onset of symptoms; however, the total window of antibody detection for IgM was only five days long (Guo et al., 2020) (See Figure 1). If the patient was not tested during the detection window, then the individual would not necessarily have a “positive” result for IgM. The authors also report the detection of IgA antibodies (median onset at five days after initial symptoms [IQR three – six days]), and 92.7% of total samples report a positive result for IgA. This same study also reports that IgG detection occurs, on average, fourteen days after initial onset of symptoms (Guo et al., 2020). Another study reports that IgA-based ELISA testing has higher sensitivity than IgG-based ELISA testing, but the IgG-based ELISA testing has higher specificity. The authors recommend IgG-based testing over the IgA-based testing in immunosurveillance studies since IgG has a longer biological half-life (Okba et al., 2020). At least one published study to date has reported that as many as 6.9% of individuals who previously had tested positive with RT-PCR results did not show the presence of antibodies for the length of the study (at least 40 days after the initial onset of symptoms) (Zhao et al., 2020).
Ideally, any rapid diagnostic test for the outpatient setting must be accurate and reliable. Current research indicates that the diagnostic window for IgA and IgM is very limited. Some data indicate that host antibody testing can also yield inaccuracies. Also, for IgG testing, the significance of positive results is questionable at the current time. A positive result could indicate a previous infection, assuming the test did not cross-react with any other IgG the host produced in response to one of the four coronaviruses known to cause the common cold in humans, for example. It is not currently known, however, if the presence of IgG antibodies indicates immunity (or degree thereof) of the host against SARS-CoV-2. The duration of any conferred immunity, or the level of IgG antibodies required to effectively acquire such immunity, are also unknown. Additional research is needed and encouraged.
Lisboa Bastos et al. (2020) performed a meta-analysis to investigate the diagnostic accuracy of serological testing for COVID-19. The authors aimed to identify studies where serological testing was compared to the “reference standard of viral culture or reverse transcriptase polymerase chain reaction.” The authors identified a total of 40 studies for inclusion in the study. The pooled sensitivity of enzyme linked immunosorbent assays (ELISAs) measuring IgG or IgM to be 84.3% (with a 95% confidence interval [CI] of 75.6% – 90.9%). For lateral flow immunoassays (LFIAs), the pooled sensitivity was found to be 66% (95% CI: 49.3% – 79.3%), and for chemiluminescent immunoassays (CLIAs), the pooled sensitivity was found to be 97.8% (95% CI: 46.2% – 100%). Pooled specificities ranged from 96.6% – 99.7%. Sensitivity was also found to be higher at least three weeks from symptom onset (69.9% to 98.9%) compared to within the first week (13.4% to 50.3%) Of the samples used to calculate specificity, 83% were “from populations tested before the epidemic or not suspected of having COVID-19.” The authors performed 49 bias risk assessments (one for methodology and one for patient selection) and identified 48 with a “high risk of patient selection bias” and 36 with “high or unclear risk of bias from performance or interpretation of the serological test.” The authors also noted that only four of the forty studies including outpatients and only two studies evaluated point-of-care testing. The authors concluded that “currently, available evidence does not support the continued use of existing point-of-care serological tests” but acknowledged that “higher quality clinical studies assessing the diagnostic accuracy of serological tests for covid-19 are urgently needed” (Lisboa Bastos et al., 2020).
Kontou et al. (2020) performed a meta-analysis investigating the use of antibody tests in detecting SARS-CoV-2. The authors focused on IgG and IgM tests based on enzyme-linked immunosorbent assays (ELISA), chemiluminescence enzyme immunoassays (CLIA), fluorescence immunoassays (FIA), and lateral flow immunoassays (LFIA). A total of 38 studies encompassing 7848 individuals (3522 COVID-19 cases, 4326 healthy controls) were included. Of the 38 studies, 21 included data for both COVID-19 cases and controls. Fourteen studies using ELISA were included, and the authors found that IgG and IgM perform “similarly” individually, but in combination, resulted in a sensitivity of 0.935. Thirteen studies using CLIA resulted in an IgG sensitivity of 0.944, an IgM sensitivity of 0.810, and a combined IgG/IgM sensitivity of 0.910. The specificities ranged from 0.954 to 0.984. Thirteen studies used LFIA and found the IgG and IgM sensitivities to range from 0.53-0.66. Combining IgG and IgM resulted in sensitivities of 0.78 – 0.83. The authors also attempted to analyze FIA-based studies but were unable to due to the paucity of studies (three identified). The authors concluded that ELISA- and CLIA-based testing performed better sensitivity-wise and that LFIA studies are “more attractive for large seroprevalence studies but show lower sensitivity” (Kontou et al., 2020).
Ko et al. (2020) investigated the differences in neutralizing antibody production between asymptomatic and “mild” symptomatic COVID-19 patients, compared to pneumonic COVID-19 patients. A total of 70 patients (15 asymptomatic, 49 mild symptomatic, and six pneumonic) were included. A microneutralization assay was performed, along with a FIA and ELISA. Neutralizing antibody production was observed in all the pneumonic patients, 93.9% of the mildly symptomatic patients, and 80% of the asymptomatic patients. Further, the entire pneumonic group showed “high” titer (defined as ≥ 1:80), while 36.7% of the mild group and 20% of the asymptomatic group showed high titer. Both the FIA (for IgG) and ELISA detected anti SARS-CoV-2 at a high sensitivity (98.8% and 97.6% respectively). The authors concluded that “Most asymptomatic and mild COVID-19 patients produced the neutralizing antibody, although the titers were lower than pneumonia patients” (Ko et al., 2020).
Wu et al. (2020) investigated the association between levels of neutralizing antibodies (NAbs) and clinical characteristics in recovered COVID-19 patients. A total of 175 patients with “mild” symptoms of COVID-19 were included. The authors found that NAbs were detected in patients starting in days 4-6 and reached peak levels in days 10 – 15. NAbs were also found not to cross-react with SARS-associated CoV, but correlated with “spike-binding antibodies targeting S1, receptor binding domain, and S2 regions. The authors also noted that NAbs titers were “significantly” higher in 56 “older” patients (1537 [IQR, 877-2427]) and 63 “middle-aged” patients (1291 [IRQ, 504 – 2126]) compared to 56 “younger patients” (459 [IQR, 225-998]). The authors concluded that “… NAb titers to SARS-CoV-2 appeared to vary substantially. Further research is needed to understand the clinical implications of differing NAb titers for protection against future infection” (Wu et al., 2020).
Kweon et al. (2020) collected 97 samples from patients with COVID-19 to analyze the serologic profiles and time kinetics of IgG and IgM against SARS-CoV-2 using the AFIAS COVID-19 Ab (BodiTechMed, 2024) and the EDI™ Novel Coronavirus COVID-19 ELISA Kit (EpitopeDiagnostics, 2024). The AFIAS assay uses recombinant nucleocapsid protein as an antigen to determine IgG and IgM antibodies against SARS-CoV-2 within 20 minutes from whole blood, serum, or plasma. The EDI™ ELISA Kit uses the microplate-based enzyme immunoassay technique to detect antibodies by measuring the optical densities (ODs) of each well of immunocomplexes. To determine the kinetics of antibodies, studies were performed at different past symptom onset (PSO) periods and to determine diagnostic accuracy of serologic assays, diagnostic sensitivity and specificities were calculated by PSO of ≤14 days and > 14 days. Kinetic studies showed that “with both assays, IgM and IgG rapidly increased after seven days post symptom onset (PSO). IgM antibody levels reached a peak at 15 – 35 d PSO and gradually decreased. IgG levels gradually increased and remained at similar levels after 22 – 35 d” (Kweon et al., 2020). The diagnostic accuracy of both serologic assays also differed based on PSO. “The sensitivity of IgG samples from ≤14 d PSO was as low as 35.7% ~ 57.1%, but it sharply increased for > 14 d PSO to 88.2% ~ 94.1%. This means that almost all patients with COVID-19 showed seroconversion after 14 d PSO, and IgG seronegative subjects in this period are considered less likely to be infected with SARS-CoV-2. In addition, both assays showed 94.2% ~ 96.4% of IgG specificities and increased IgG titers in COVID-19 patients were maintained. Thus, IgG serologic assays can be useful for ruling out SARS-CoV-2 infection after 14 d PSO, detecting past infection, and epidemiologic surveys” (Kweon et al., 2020). For IgM, the sensitivities were “as low as 21.4% (same in both assays) in the samples collected ≤ 14 d PSO and 41.2% ~ 52.9% in samples > 14 d PSO. These findings indicated that in patients infected with SARS-CoV-2, IgM seroconversion may not develop or might not be detected until the middle or late stages of infection. In other words, SARS-CoV-2 infection may be missed based on IgM seropositivity; thus, IgM tests must not be solely used in COVID-19 diagnosis and should be used only as a supportive tool in addition to molecular tests” (Kweon et al., 2020). In addition, IgM titers in COVID-19 patients showed a significant reduction after 35 d PSO; therefore, their utility in detecting past infection is limited. The author concludes that “testing for antibodies against SARS-CoV-2, especially IgG, has the potential for ruling out SARS-CoV-2 infection after 14 d PSO, detecting past infection, and epidemiologic surveys” (Kweon et al., 2020).
Caturegli et al. (2020) performed a case-control study to determine the clinical utility and validity of using SARS-CoV-2 antibodies, which were serum IgG and IgA antibodies formed against the SARS-CoV-2 spike protein detected by enzyme-linked immunosorbent assay (ELISA). When assays were formed 14 days or later after symptom onset, the researchers found that the sensitivity was 0.976 (95% CI, 0.928 to 0.995) and specificity was 0.988 (95% CI, 0.974 to 0.995), but the sensitivity decreased at earlier time points. Antibodies “predicted the odds of developing acute respiratory distress syndrome, which increased by 62% (CI, 48% to 81%; P < 0.001) for every 2-fold increase in IgG.” This demonstrates the linkage of antibodies used to measure clinical severity and for those who tested negative by NAAT but remained potentially COVID-positive.
In a household cohort study, Churiwal et al. (2021) assessed the utility of a rapid point of care test for COVID-19 antibodies by comparing the performance of BioMedomics COVID-19 IgM/IgG Rapid Antibody Test against an ELISA. The test was performed on 303 patients at study enrollment and four weeks later. According to the results, sensitivity was lower early in infection and those who never developed symptoms (74% sensitivity). Only two were detected among 499 tests early in infection due to false-positive IgM bands. When measured four weeks later after the onset of symptoms, it demonstrated robust sensitivity (90%) and complete specificity (100%). The authors conclude that "When used appropriately, rapid antibody tests offer a convenient way to detect symptomatic infections during convalescence” (Churiwal et al., 2021).
Fox et al. (2022) performed a meta-analysis to assess the accuracy of antibody tests. The analysis covered 178 studies with a total of 64,688 samples taken from 25,724 people with confirmed SARS-CoV-2. All the studies were conducted before the introduction of the SARS-CoV-2 vaccines to ensure the responses were due to naturally acquired antibodies. The average sensitivity for either IgG or IgG combined with IgM was 41.1% one week after symptom onset, 74.9% two weeks after symptom onset, and 88.0% three weeks after symptom onset. The average sensitivity during the convalescent phase of infection, up to 100 days since symptom onset, was 89.8% for IgG, 92.9% for IgG or IgM combined, and 94.3% for total antibodies. The average sensitivities for IgM alone “followed a similar pattern but were of a lower test accuracy in every time slot.” The authors conclude that antibody tests “could be a useful diagnostic tool” but note that “antibody tests have an increasing likelihood of detecting an immune response to infection as time since onset of infection progresses and have demonstrated adequate performance for detection of prior infection for sero-epidemiological purposes” and “the applicability of results for detection of vaccination-induced antibodies is uncertain” (Fox et al., 2022).
Antigen Testing
Another possible diagnostic testing methodology is antigen detection testing, which relies upon the direct detection of parts of the virus called “antigens”—in this instance, proteins located on the outside of SARS-CoV-2, such as the spike protein (S) or nucleocapsid protein, that can cause an immune response in an individual. What makes this method of testing distinct from antibody testing is that antigen testing directly measures the presence of the virus in a person whereas antibody testing is measuring the patient’s response to an infection. These antigen detection tests can be deployed as rapid antigen tests that decrease the turnaround time for results but usually lack specificity (Loeffelholz & Tang, 2020).
On May 8, 2020, the FDA issued the first EUA for antigen testing for COVID-19 to the Quidel Corporation for their Sofia®2 SARS Antigen FIA lateral flow immunofluorescent sandwich assay for the qualitative detection of the nucleocapsid (N) protein antigen of SARS-CoV-2 for use in individuals suspected of COVID-19 by their healthcare provider (Quidel Corporation, 2020). This test has been approved as a point-of-care (POC) test (FDA, 2024c). This test functions by detecting the N protein of either the SARS-CoV or SARS-CoV-2 virus from an upper respiratory sample (either a nasal swab or nasopharyngeal swab). First, the sample is placed in a reagent tube so that any virus, if present, is broken apart to allow for the N proteins to be exposed. The sample then travels from the sample well, down a test strip — where the term “lateral flow” is derived — where the proprietary reagents will recognize any N proteins and trap them in place on the strip. The test requires at least 15 minutes to develop prior to analysis. The strip can then be read by the Sofia®2 system that measures the fluorescent signal from the proprietary reagents. The Sofia®2 system allows the user to have two different modes for analysis—“Walk Away” and “Read Now.” For the “Walk Away” mode, the user will insert the test cassette strip into the system, and the results will be displayed in 15 minutes because the test will be developed while in the instrument. In “Read Now” mode, the user must have already allowed at least 15 minutes for the test to develop prior to inserting it into the instrument. Then, the Sofia®2 system will display the result within one minute (Quidel Corporation, 2020). On Aug. 20, 2020, Quidel reported that the Sofia test’s labeling had been amended to include “either nasal or nasopharyngeal swabs” thereby allowing Quidel a second corresponding kit configuration (BioSpace, 2020).
On July 2, 2020, a second antigen test (BD Veritor System for Rapid Detection of SARS-CoV-2) from Becton, Dickinson, and Company was issued an EUA. This test is described as “a chromatographic digital immunoassay intended for the direct and qualitative detection of SARS-CoV-2 nucleocapsid antigens in nasal swabs from individuals who are suspected of COVID-19 by their healthcare provider within the first five days of the onset of symptoms.” The test is authorized for use in POC settings. The test’s mechanism of action is as follows: if there are any antigens in the sample (in this case, the nucleocapsid of the virus), they will bind to antibodies conjugated to detector particles in the test strip. The new “conjugates” migrate to the “reaction area” and are captured by another line of antibodies. The test reads positive when the conjugate is found at both “Control” and “Test” positions on the device. BD Veritor reported the following values for the test (in comparison to RT-PCR): 84% positive predictive agreement, 100% negative predictive agreement, 98% overall percent agreement, 100% positive predictive value, and 97.5% negative predictive value. No cross-reactivity was reported (BD Veritor, 2020).
On August 18, 2020, a third antigen test (LumiraDx SARS-CoV-2 Ag Test from LumiraDx UK Ltd.) was issued an EUA. The test is described as “a single use fluorescence immunoassay device designed to detect the presence of the nucleocapsid protein antigen directly from SARS-CoV-2 in nasal swab specimens, without transport media.” The mechanism of action is as follows: when a droplet of the specimen is added to the “Test Strip,” pre-made reagents on the strip react with any antigen in the specimen. The amount of fluorescence created is proportional to the amount of antigen detected. LumiraDx reported a limit of detection of 32 TCID50/mL [tissue culture infectious dose], as well as a 97.6% positive percent agreement, 96.6% negative percent agreement, 93.1% positive predictive value, 98.8% negative predictive value, and 96.9% overall percent agreement (based on 257 total samples) (LumiraDx, 2020).
As of April 20, 2022, 50 antigen tests have Emergency Use Authorization (EUA) by the U.S. Food and Drug Administration (FDA) (FDA, 2023a). These testing methods include (among others): Bulk Acoustic Wave (BAW) Biosensors, Chemiluminescence Immunoassays, Chromatographic Digital Immunoassays, Digital Lateral Flow, Magnetic Force-assisted Electrochemical Sandwich Immunoassay (MESIA), Microfluidic Immunofluorescence Assay, and Paramagnetic Microbead-based Immunoassay (FDA, 2023a).
Clinical Utility and Validity of Antigen Testing
To address the clinical performance, two primary studies were performed. Both studies only used frozen samples. The first study used 143 samples with 80% PPA or Positive Percent Agreement (47/59 of positive samples tested “positive”). They report 100% NPA or Negative Percent Agreement — all 84 negative samples tested “negative.” The second study used a total of 48 samples. Again, 80% of the positive samples tested “positive;” however, only a total of five positive samples were included within this second study. The remaining 43 samples were all negative samples. This study reports a sensitivity of 80.0%, but a 95% confidence interval range of 37.6% – 96.4%. A third supportive study was also performed. In this study, thirty swabs were taken. Twenty of these swabs were spiked with one lower concentration of the virus while the remaining ten swabs were spiked with a higher concentration of the virus. Then, all 30 swabs were tested and compared to 47 control (“unspiked”) samples. In this study, none of the “unspiked” control samples tested “positive” while all 30 of the “spiked” samples, regardless of the concentration, tested positive. Quidel also tested the LoD of the Sofia®2 SARS Antigen FIA test. LoD is typically measured by determining the TCID50 (median tissue culture infective dose). The TCID50 is the amount where 50% of the cells within a sample are infected (Wulff et al., 2012). For the Sofia®2 SARS Antigen FIA test, the LoD for a direct swab sample has a TCID50 of 113 mL whereas it is 850 mL if the initial sample is from a swab sample that has been diluted into three mL of reagent. Finally, Quidel also checked this antigen test for possible cross-reactivity with several microorganisms and other viruses. It shows no cross-reactivity with any of the microorganisms or viruses tests other than SARS-CoV. Of note, it does not cross-react with human coronavirus 229e, OC43, NL63, or MERS-CoV (heat-inactivated); however, they did not check for possible cross-reactivity with the other known human coronavirus (HKU1) due to a lack of availability at this time. This is noteworthy since this coronavirus is associated with the common cold. Limitations of the Sofia®2 SARS Antigen FIA test includes the following:
- This test must be performed using the Sofia®2 system, and the test must be performed accurately following the test procedure. Failure to do so can adversely affect the performance of the test and may invalidate the results.
- A positive test cannot distinguish between a SARS-CoV or a SARS-CoV-2 infection. SARS-CoV is the virus that caused the SARS outbreak of 2003. It should be noted that there is no current outbreak of SARS.
- This test also does not distinguish between “live” (viable) virus and non-viable virus. Consequently, the test results do not necessarily correlate with viral culture results performed on the same sample.
- This test is only for the qualitative use on a sample from either a nasal swab or a nasopharyngeal swab. It has not been approved for use, at this time, on any other sample, such as saliva.
- Negative test results can occur if the viral level is below the lower limit of the test. All negative results “should be treated as presumptive and confirmed with an FDA authorized molecular assay, if necessary, for clinical management, including infection control” (Quidel Corporation, 2020).
- Positive test results do not rule out coinfections, and negative results do not “rule in” other non-SARS viral or bacterial infections.
- The clinical performance assays submitted for FDA approval were performed using frozen samples; the test may have a different performance when used with a fresh sample (such as in a point-of-care setting).
- “If the differentiation of specific SARS viruses and strains is needed, additional testing, in consultation with state or local public health departments is required” (Quidel Corporation, 2020).
- As previously noted, the company did not check this test (as of publication date) for cross-reactivity with human coronavirus HKU1 due to a lack of availability of that strain. This is notable since this virus is associated with upper respiratory conditions such as the common cold.
One multi-center study, currently a preprint at the time of publication, reports the development of another rapid antigen detection test (RADT) that screens for SARS-CoV-2 by targeting the nucleocapsid protein. This test, when using a nasopharyngeal swab sample, reports a 100% positive agreement with RT-PCR testing. They also report 73.6% positive agreement when using a urine sample (Diao et al., 2020). This study is yet to be published in a peer-reviewed journal, and the test is not FDA approved as of May 18, 2020. Another study published recently in ACS Nano reports on the development of a RADT using field-effect transistor (FET)-based biosensing where a graphene sheet for the FET is coated with a specific antibody against the SARS-CoV-2 spike protein. This method can detect the protein in concentrations as low as one fg/mL in buffer and has an LOD of 242 copies/mL for a clinical sample (versus 16/mL for a culture medium) (Seo et al., 2020). To date, the WHO states that “Ag-RDTs could play a significant role in guiding patient management, public health decision making and in surveillance of COVID-19. Currently, there is insufficient evidence on performance and operational use to recommend specific commercial products” (WHO, 2021a).
Scohy et al. (2020) evaluated the Coris COVID-19 Ag [Antigen] Respi-Strip test in comparison to RT-PCR. The authors tested 148 nasopharyngeal swabs, with 106 testing positive by RT-PCR. The rapid antigen test detected 32 of these 106 positive results, for a sensitivity of 30.2%. All samples deemed positive by the antigen test were also deemed positive by RT-PCR. The authors noted that higher viral loads were associated with better detection by antigen tests but concluded that “the overall poor sensitivity of the COVID-19 Ag Respi-Strip does not allow using it alone as the frontline testing for COVID-19 diagnosis” (Scohy et al., 2020).
Mak et al. (2020) evaluated the BIOCREDIT COVID-19 Ag test in comparison to RT-PCR. The BIOCREDIT test’s limit of detection (LOD) was compared to RT-PCR and viral culture, and a total of 368 samples from confirmed COVID-19 cases were included. A sample volume of 100 μL was used. The authors found the LOD of BIOCREDIT to be 1000-fold less sensitive than viral culture (BIOCREDIT LOD: 10-2, viral culture: 10-5). RT-PCR’s LOD was measured to be 10-7. Further, BIOCREDIT detected between 11.1% and 45.7% of RT-PCR positive patients from COVID-19 patients. The authors concluded that “This study demonstrated that the RAD test serves only as adjunct to RT-PCR test because of potential for false-negative results” (Mak et al., 2020).
Lambert-Niclot et al. (2020) analyzed the COVID-19 Ag Respi-Strip test and compared its accuracy to RT-PCR. A total of 138 nasopharyngeal samples were included, with 94 testing positive by RT-PCR. The Respi-Strip test identified 47 of 94 positive specimens for a sensitivity of 50%, although the specificity was 100% for both tests. The authors also noted that the control lines were “barely” visible for 17 tests (nine positive and eight negative). The authors acknowledged that due to the low prevalence in France (the country in which this study was performed), prospective studies should be undertaken(Lambert-Niclot et al., 2020).
Hirotsu et al. (2020) evaluated a new antigen test (LUMIPULSE) which is based on chemiluminescence enzyme immunoassay. A total of 313 nasopharyngeal swabs were included (82 serial samples from seven COVID patients, 231 individual samples from four COVID patients and 215 healthy controls). These samples were tested by both LUMIPULSE and RT-PCR. Compared to RT-PCR, LUMIPULSE demonstrated a 91.4% overall agreement rate (286/313), with a 55.2% sensitivity and 99.6% specificity. At >100 viral copies, LUMIPULSE agreed perfectly with RT-PCR, and at 10-100 viral copies, there was an 85% concordance rate (with concordance declining at lower viral loads). The authors concluded that “the LUMIPULSE antigen test can rapidly identify SARS-CoV-2-infected individuals with moderate to high viral loads and may be helpful for monitoring viral clearance in hospitalized patients” (Hirotsu et al., 2020).
Villaverde et al. (2021) conducted a multicenter study to compare the diagnostic accuracy of the Panbio coronavirus disease 2019 Antigen Rapid Test of nasopharyngeal samples in pediatric patients with COVID-19 symptoms ≤ 5 days. They demonstrated “limited accuracy in nasopharyngeal antigen testing: overall sensitivity was 45.4%, and 99.8% of specificity, positive-predictive value was 92.5%,” with moderate concordance between the RT-PCR and antigen test. They noted that a high proportion of false-negative results from the antigen tests (54.5%) may have public health implications in unknown spreading of the virus. But because this test has a good positive likelihood ratio, and is cheap, rapid, and widely distributed, it may be used as a first screening test in a pandemic situation, though its value as a diagnostic tool is questionable due to the low sensitivity and negative likelihood ratio.
Peacock et al. (2022) studied the clinical utility of the BinaxNOW antigen test by Abbott Diagnostics, a lateral flow immunochromatographic point-of-care test which provides results in 15 minutes from a nasal swab. BinaxNOW was performed on 735 samples and results were compared to PCR. In total, 623 of 735 (84.8%) had symptoms and 460 of 623 patients (62.6%) had symptoms for less than seven days. Positive 141 (19.2%) with the BinaxNOW test. Those with symptoms for more than two weeks had a positive test rate half of those with earlier onset. "In patients with symptoms ≤7 days, the sensitivity, specificity, and negative and positive predictive values for the BinaxNOW test were 84.6%, 98.5%, 94.9%, and 95.2%, respectively" (Peacock et al., 2022). The authors conclude that BinaxNOW has good sensitivity and specificity and is recommended for patients with symptoms up to two weeks (Peacock et al., 2022).
Panel Testing
Multiple laboratories have developed panels to screen for possible microorganism infections from a single sample. For example, multiplex PCR can simultaneously detect multiple pathogens rather than sequentially testing for each individual pathogen. Such testing can be advantageous when different pathogens may manifest with similar clinical presentation; however, this testing can be costly and can also result in false-negatives if preferential amplification of one target over another occurs. As of May 4, 2022, the BioFire® Respiratory Panel 2.1 (RP2.1), the QIAstat-Dx® Respiratory SARS-CoV-2 Panel, ePlex Respiratory Pathogen Panel 2, cobas SARS-CoV-2 & Influenza A/B, Xpert Xpress SARS-CoV-2/Flu/RSV, Quest Diagnostics RC COVID-19 +Flu RT-PCR, Sofia 2 Flu + SARS Antigen FIA, and the Influenza SARS-CoV-2 (Flu SC2) Multiplex Assay from the CDC received an EUA from the FDA for testing for COVID-19 (FDA, 2024c). The BioFire® Respiratory Panel 2.1, the QIAstat-Dx® Respiratory SARS-CoV-2 Panel, and ePlex Respiratory Pathogen Panel 2 use multiplex nucleic acid testing from a nasopharyngeal swab to detect and differentiate microorganisms listed in Table 1 (BioFire, 2020; GenMark Diagnostics, 2024; Qiagen GmbH, 2021), whereas the CDC Multiplex detects and differentiates influenzas A and B from SARS-CoV-2 (FDA, 2021c).
| Table 1: Respiratory Pathogen Panel Testing Containing SARS-CoV-2 |
||
| BioFire® Respiratory Panel 2.1 |
QIAstat-Dx® Respiratory SARS-CoV-2 Panel |
ePlex Respiratory Pathogen Panel 2 |
|
|
H1-2009
|
Clinical Utility and Validity of Panel Testing
The BioFire RP2.1 panel must be used with either the BioFire FilmArray 2.0 or BioFire FilmArray Torch Systems, and it does not provide a quantitative value for any organism within the sample. This panel “has not been established for specimens collected from individuals without signs or symptoms of respiratory infection” (BioFire, 2020). This panel has not been validated for the monitoring of treatment for any condition. If a test result shows four or more organisms detected, then the sample should be retested. A negative result does not necessarily exclude an infection. “Negative test results may occur from the presence of sequence variants (or mutation) in the region targeted by the assay, the presence of inhibitors, technical error, sample mix-up, an infection caused by an organism not detected by the panel, or lower respiratory tract infection that is not detected by a nasopharyngeal swab specimen” (BioFire, 2020).
The BioFire RP2.1 panel cannot necessarily distinguish between existing viral strains and new variants. One example is the inability to distinguish between Influenza A H3N2v and seasonal Influenza A H3N2. This panel also cannot reliably differentiate between human rhinovirus and enterovirus due to genetic similarity. If detected, the “result should be followed-up using an alternate method (e.g., cell culture or sequence analysis) if differentiation between the viruses is required” (BioFire, 2020). The performance characteristics of several microorganisms detected by this panel, including HCoV 229E, were determined using retrospective clinical specimens due to the small number of positive specimens collected. The BioFire RP2.1 panel should not be used if B. pertussis is suspected because of its low sensitivity. “[A] B. pertussis molecular test that is FDA-cleared for use on patients suspected of having a respiratory tract infection attributable to B. pertussis only should be used instead” (BioFire, 2020). This is because the RP2.1 panel targets a single-copy promoter target (ptxP) whereas more sensitive tests target the multi-copy IS481 insertion sequence. The BioFire RP2.1 panel also shows cross-reactivity with B. bronchiseptica and B. parapertussis at higher concentrations.
The primers used in the BioFire RP2.1 panel to detect COVID-19 may cross-react with coronaviruses from other species due to high sequence homology. BioFire reports predicted cross-reactivity with up to three bat coronaviruses (accession: MN996532, MG772933, and MG772934) and one pangolin coronavirus (accession: MT084071). However, “[i]t is unlikely that these viruses would be found in a human clinical nasopharyngeal swab; but if present, the cross-reactive product(s) produced by the BioFire RP2.1 will be detected as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)” (BioFire, 2020).
The difference between the BioFire RP2 panel and the BioFire RP2.1 panel is the ability to detect SARS-CoV-2. Consequently, within the Instructions for Use (IFU) for the RP2.1 panel, BioFire reports on the data of the RP2 panel. The clinical performance of the RP2 panel was determined using both fresh and frozen samples. The clinical performance values for the four endemic HCoVs are listed in Table 2 (BioFire, 2020). They note a cross-reactivity between HCoV-OC43 and HCoV-HKU1.
| Table 2: Clinical Performance of BioFire RP2/RP2.1 Panel for Endemic HCoVs |
||||
| Analyte |
PPA |
PPA 95% CI |
NPA |
NPA 95%CI |
| HCoV-229E |
11/12 (91.7%) |
64.6 – 98.5 |
1595/1600 (99.7%) |
99.3 – 99.9 |
| HCoV-HKU1 |
43/43 (100%) |
91.8 – 100 |
1557/1569 (99.2%) |
98.7 – 99.6 |
| HCoV-NL63 |
40/40 (100%) |
91.2 – 100 |
1562/1572 (99.4%) |
98.8 – 99.7 |
| HCoV-OC43 |
33/41 (80.5%) |
66.0 – 89.8 |
1566/1571 (99.7%) |
99.3 – 99.9 |
| Notes: Abbreviations used—PPA (Positive Percent Agreement); NPA (Negative Percent Agreement). |
||||
Concerning the detection of SARS-CoV-2, the BioFire RP2.1 panel reports a limit of detection (LoD), using the USA-WA1/2020 isolate, of 500 copies/mL when using a heat-inactivated virus. They report a 100% detection rate (20/20). This equates to 6.9 X 10-2 TCID50/mL. They also tested the LoD using an infectious virus isolate obtained from the World Reference Center for Emerging Viruses and Arboviruses, contributed by the CDC. With this infectious sample, the LoD was determined to be 160 copies/mL (or 1.1 X 10-2 TCID50/mL). Again, they report a 100% detection rate (20/20) (BioFire, 2020).
Similar to the BioFire panel test, the QIAstat-Dx Respiratory SARS-CoV-2 panel test by Qiagen is for use on a proprietary system, the QIAstat Dx Analyzer System. It is also a qualitative test approved for testing in “patients suspected of COVID-19 by their healthcare provider.” It is also “not intended to be used as the sole basis for diagnosis, treatment, or other patient management decisions” (Qiagen GmbH, 2021). It is important to note that the test performance in either immunocompromised individuals or asymptomatic individuals has not been established as of publication date. A positive test result cannot rule out a co-infection; an erroneous negative test result can be due to erroneous sample handling as well as variations in the target sequences, organism levels below the limits of detection, and/or use of an interfering reagent (such as certain medications or therapies). Since the QIAstat-Dx test targets the E gene of SARS-CoV-2, which is homologous to sequences in multiple bat SARS viruses, it is possible to cross-react with these bat SARS viruses; however, the likelihood of infection of these viruses in humans is unlikely since none have been reported to date (Qiagen GmbH, 2021).
Also, like the BioFire RP2/RP2.1 panel tests, the QIAstat-Dx test may not distinguish between existing viral strains and emerging viral strains, such as influenza A. However, unlike the BioFire RP2/RP2.1 panel tests, the QIAstat-Dx test does detect the IS481 multi-copy insertion sequence present in multiple Bordetella species. This does increase the sensitivity of the test, but it can increase the possibility of false-positive results if the specimen is contaminated with a non-pertussis Bordetella species (Qiagen GmbH, 2021).
In addressing the clinical performance of the QIAstat-Dx test for detecting SARS-CoV-2, Qiagen set up two positive trials (one at a higher concentration sample [n = 10] and one at a low positive contrived sample [n = 20), and they report a positive percent agreement (PPA) of 100% (30/30) (95% CI: 85.8% – 100%). Likewise, they did a negative control (n = 30) and report a negative percent agreement (NPA) of 100% (30/30) (95% CI: 85.8% – 100%). In reporting the limit of detection (LoD), they used 20 replicates with a detection rate of at least 95% (or 19/20) to generate a ‘positive’ signal. Using source material obtained from the clinical sample strain of the Hospital of Barcelona (Spain), Qiagen reports an LoD of 500 copies/mL.
The performance of the other targets within the panel were assessed in a multi-center study conducted at six geographically diverse study sites—Copenhagen, Denmark; Minneapolis, MN; Indianapolis, IN; Liverpool, NY; Columbus, OH; and Albuquerque, NM. The performance was determined using both frozen and fresh samples. The clinical performance values for the four endemic HCoVs are listed in Table 3 (Qiagen GmbH, 2021).
| Table 3: Clinical Performance of QIAstat-Dx Panel for Endemic HCoVs |
||||
| Analyte |
PPA |
PPA 95% CI |
NPA |
NPA 95%CI |
| HCoV-229E |
8/9 (88.9%) |
56.5 – 98.0 |
1975/1975 (100%) |
99.8 – 100.0 |
| HCoV-HKU1 |
51/52 (98.1%) |
89.9 – 99.7 |
1925/1932 (99.6%) |
99.3 – 99.8 |
| HCoV-NL63 |
40/47 (85.1%) |
72.3 – 92.6 |
1936/1938 (99.9%) |
99.6 – 100.0 |
| HCoV-OC43 |
26/29 (89.7%) |
73.6 – 96.4 |
1951/1955 (99.8%) |
99.5 – 99.9 |
| Notes: Abbreviations used—PPA (Positive Percent Agreement); NPA (Negative Percent Agreement). |
||||
As with the other two tests, the ePlex RP2 Panel “should not be used as the sole basis for diagnosis, treatment, or other patient management decisions. Positive results are indicative of active infection with the identified respiratory pathogen but do not rule out infection or co-infection with non-panel organisms. The agent detected by the ePlex RP2 Panel may not be the definite cause of disease. Negative results for SARS-CoV-2 and other organisms on the ePlex RP2 Panel may be due to infection with pathogens that are not detected by this test, or lower respiratory tract infection that may not be detected by a nasopharyngeal swab specimen. Negative results do not preclude infection with SARSCoV-2 or other organisms on the ePlex RP2 Panel and should not be used as the sole basis for patient management decisions. Negative results must be combined with clinical observations, patient history, and epidemiological information” (GenMark Diagnostics, 2024). A limitation of ePlex RP2 Panel is its unpredictability in differentiating human rhinovirus and enterovirus due to genetic similarity. If differentiation is required, an ePlex RP2 Panel positive human rhinovirus/enterovirus result should be followed up using an alternative method, such as cell culture or sequence analysis. Cross-reactivity with SARS-CoV-1 is also observed at high titers.
To test the performance characteristics of ePlex RP2 Panel for SARS-CoV-2 detection, 170 nasopharyngeal previously frozen swab samples were collected (59 known SARS-CoV-2 positive and 111 presumed SARSCoV-2 negative samples). “Positive percent agreement (PPA) was calculated by dividing the number of true positive (TP) results by the sum of TP and false negative (FN) results, while negative percent agreement (NPA) was calculated by dividing the number of true negative (TN) results by the sum of TN and false positive (FP) results” (GenMark Diagnostics, 2024). The ePlex RP2 Panel detected SARS-CoV-2 in 59/59 positive specimens (100% positive percent agreement) and confirmed 111/111 negative specimens (100% negative percent agreement). To determine the limit of detection (LoD), the lowest concentration at which SARS-CoV-2 is detected at least 95% of the time, serial dilutions were prepared in a natural clinical matrix and at least 20 replicates per concentration were tested in the study. “The LoD concentration for detection of SARS-CoV-2 was determined to be 0.01 TCID50/mL, which corresponds to 250 genomic copies per milliliter, as determined by digital droplet PCR” (GenMark Diagnostics, 2024).
Regarding the “Influenza SARS-CoV-2 (Flu SC2) Multiplex Assay” from the CDC, the FDA reported a limit of detection (LOD) of 1.01 x 10-2 (at ID50 [infective dose] / reaction). The panel was evaluated using 104 samples (33 positive for SARS-CoV-2, 30 positives for influenza A, and 30 positives for influenza B, 11 negative samples), and compared to an RT-PCR assay. There was a 100% concordance rate between the two tests. Additionally, cross-reactivity between the three analytes and 35 common respiratory pathogens (16 viruses, 18 bacterial species, one yeast) was evaluated, and no cross-reactivity was identified (FDA, 2024b).
The cobas SARS-CoV-2 & Influenza A/B panel is approved for emergency use authorization by the FDA; the panel uses qualitative detection of nucleic acids from SARS-CoV-2 in pooled samples. Six cultured viruses are tested for, two each of influenza A and influenza B strains as well as SARS-CoV-2. In an independent study, Poljak et al. (2020) performed a clinical evaluation of the cobas SARS-Cov-2 test (non-inclusive of influenza A/B panel). The cobas SARS-CoV-2 test was evaluated against an in house and well-characterized comparator using 217 samples. cobas and the comparator showed overall agreement of 98.1%. Another comparative evaluation on 502 samples showed agreement of 99.6%. The authors concluded that cobas “is a reliable assay for qualitative detection of SARS-CoV-2 in nasopharyngeal swab samples collected in the Universal Transport Medium System (UTM-RT)” (Poljak et al., 2020).
There are other panels that are not yet FDA approved such as the AMPLIQUICK® Respiratory Triplex assay that detects and differentiates between SARS-CoV-2, influenza A/B and respiratory syncytial viruses in respiratory specimens. Results from AMPLIQUICK® were compared to the Allplex™ Respiratory Panel 1 and 2019-nCoV assays. A total of 359 predetermined respiratory samples with diagnosed SARS-CoV-2, influenza A, influenza B and RSV were included in the study. The AMPLIQUICK® Respiratory Triplex “showed high concordance with the reference assays, with an overall agreement for SARS-CoV-2, influenza A, influenza B, and RSV at 97.6%, 98.8%, 98.3% and 100.0%, respectively.” The authors conclude that the "AMPLIQUICK® Respiratory Triplex is a reliable assay for the qualitative detection and differentiation of SARS-CoV-2, influenza A, influenza B, and RSV in respiratory specimens, which may prove useful for streamlining diagnostics during the winter influenza-seasons" (Mboumba Bouassa et al., 2022).
Miscellaneous Testing
Other methodologies have been proposed to complement or even replace the standard tests described above. For example, a new “RT-LAMP” (reverse transcription loop-mediated isothermal amplification) application has started to see some use for the COVID-19 pandemic. This technique attempts to combine the speed of antigen testing and the accuracy of nucleic acid testing; RT-LAMP includes the traditional reverse transcriptase (RT), as well as a DNA polymerase with “strong strand displacement activity and tolerance for elevated temperatures and up to six DNA oligonucleotides of a certain architecture.” These oligonucleotides act as primers for the RT, but additional oligonucleotides for the DNA polymerase are designed so that the DNA products loop back into their ends. This results in “self-priming templates” for the DNA polymerase, which allows the reaction [the nucleic acid amplification] to proceed as normal. Detection of the amplified DNA without specialized instrumentation is the key challenge; some tests use a pH indicator that changes the color of the solution the reaction is run in. Since the reaction does not require the use of a thermal cycler with real time fluorescence measurement, the results can be delivered in a faster time frame than traditional RT-PCRs (Dao Thi et al., 2020).
Nagura-Ikeda et al. (2020) evaluated the “clinical performance of six molecular diagnostic tests and a rapid antigen test for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).” Self-collected saliva was the medium used for analysis. A total of 103 patients with COVID-19 were included (15 asymptomatic, 88 symptomatic). The six molecular diagnostic tests included three RT-PCR tests, an RT-qPCR test, a “cobas SARS-CoV-2 high-throughput system” and an RT-LAMP assay. The molecular diagnostic tests detected viral RNA in 50.5%-81.6% of specimens and an antigen was detected in 11.7% of the specimens by the rapid antigen test. Viral RNA was also detected at a higher rate (65.6% – 93.4%) in specimens collected within nine days of symptom onset compared to specimens collected after 10 days (22.2% – 66.7%). Viral RNA was detected in asymptomatic patients at a rate of 40% – 66.7%. The authors concluded “Self-collected saliva is an alternative specimen option for diagnosing COVID-19. LDT RT-qPCR…and RT-LAMP showed sufficient sensitivity in clinical use to be selectively used according to clinical settings and facilities. The rapid antigen test alone is not recommended for initial COVID-19 diagnosis because of its low sensitivity” (Nagura-Ikeda et al., 2020).
Dao Thi et al. (2020) performed a validation of a “two-color RT-LAMP assay protocol for detecting SARS-CoV-2 viral RNA using a primer set specific for the N gene.” The authors wrote that a positive sample would be detected by a color change from red to yellow and tested their RT-LAMP assay on “surplus RNA samples isolated from 768 pharyngeal swab specimens collected from individuals being tested for COVID-19.” The results were compared to a traditional RT-qPCR assay. The specificity of the RT-LAMP assay was found to be 99.7%. Further, the RT-qPCR positive samples with a cycle threshold (CT) number of under 30 scored positive (agreeance) in the RT-LAMP assay at a 97.5% agreeance rate. Agreeance rate declined both at the 30 – 35 threshold and at the 35 – 40 threshold. The authors also developed a “swab-to-RT” LAMP protocol, which was measured at 86% sensitivity (for CT < 30) and a 99.5% specificity. The authors concluded that “The RT-LAMP assay and LAMP-sequencing extend the range of available test methods and complement individual tests and pooled tests based on RT-qPCR with a faster, simpler, and potentially more cost-effective test method” (Dao Thi et al., 2020).
R. Wang et al. (2020) demonstrated a one-pot visual SARS-CoV-2 detection system named “opvCRISPR” by integrating reverse transcription loop-mediated isothermal amplification (RT-LAMP) and Cas12a cleavage in a single reaction system, which simplifies operations and avoids contamination. The opvCRISPR enables detection at every single molecular level in forty-five minutes. “The RT-LAMP reagents are incubated at the bottom of the tube, and CRISPR/Cas12a reaction reagents are added on the lid. SARS-CoV-2 RNA templates extracted from the respiratory swab are amplified by RT- LAMP, followed by mixing with the Cas12a reagents for cleavage. Once the Cas12a nuclease is activated by recognizing DNA target, it splits the quenched fluorescent single-stranded DNA (ssDNA) reporter (FAM- TTATT-BHQ1) indiscriminately, generating the fluorescence signal visible to the naked eye under blue light” (R. Wang et al., 2020). To investigate the diagnostic accuracy of opvCRISPR, 26 SARS-CoV-2 RT-PCR positive respiratory swab samples and 24 SARS-CoV-2 RT-PCR negative samples were tested. “All infected samples were determined to be SARS-CoV-2 positive while all uninfected samples tested to be negative by both opvCRISPR and RT- PCR. The opvCRISPR diagnostic results provide 100% agreement with the Centers for Disease Control and Prevention (CDC)-approved quantitative RT-PCR assay” (R. Wang et al., 2020). The author states that “the proposed method only requires minimal equipment, demonstrating great potential in enabling next-generation molecular diagnosis towards point-of- care diagnosis. However, the present method requires additional step to extract RNA. Further efforts need to be made to combine the RNA extraction module with the opvCRISPR to achieve from sampling to result nucleic acid detection” (R. Wang et al., 2020).
Another methodology with potential application for COVID-19 testing is next-generation sequencing (NGS). The NGS procedure typically includes the following steps: first the patient’s DNA is prepared to serve as a template, then DNA fragments are isolated (on solid surfaces such as small beads) where sequence data is generated, then these results are compared against a reference genome. Any DNA sample may be used if the quality and quantity of that sample are sufficient, but the methods of library generation and data analysis often vary from panel to panel. NGS is often used to produce swift and high-volume sequencing (Hulick, 2024). The FDA issued an EUA to Illumina, Inc. for the Illumina COVIDSeq Test on June 10, 2020 but has since updated its indications on October 29, 2020 to be for the “qualitative detection of SARS-CoV-2 RNA from nasopharyngeal (NP) swabs, oropharyngeal (OP) swabs, anterior nasal swabs, mid-turbinate nasal swabs, nasopharyngeal wash/aspirates, nasal aspirates, and bronchoalveolar lavage (BAL) specimens from individuals suspected of COVID-19 by their healthcare provider” (FDA, 2021b). The FDA also issued an EUA to Helix OpCo LLC (dba Helix) for the Helix COVID-19 NGS Test on August 6, 2020. The test detects the gene for the SARS-CoV-2 spike protein, as well as one internal control (the human gene RPP30). The limit of detection was found to be 125 genetic copy equivalents / mL, and both the positive and negative percent agreements were measured to be 100% over 30 samples (Helix, 2020).
Furthermore, whole genome sequencing (WGS) has been demonstrated to have application for COVID-19 testing as well. WGS is conducted through four steps of DNA shearing, by using “molecular scissors” to cut DNA; then DNA bar-coding, for which “scientists add small pieces of DNA tags, or bar codes to identify which piece of sheared DNA belongs to which [pathogen];” then the bar-coded DNA is put into the whole genome sequencer that identifies the bases; and finally, the data is analyzed to compare sequences and identify possible differences (CDC, 2024b). In several countries, like the Netherlands, China, Vietnam, and the United States, particularly rapid WGS has been beneficial in informing outbreak response, general public health decision making, and infection risk in various facilities (Chau et al., 2021; Oude Munnink et al., 2020; Taylor et al., 2020; F. Wang et al., 2020). In the Netherlands, WGS with the first cases in February 2020 was able to confirm separate introductions of the virus into the country, and attribute increases in case prevalence to co-circulating virus variants following the spring holidays. WGS informed the sequence diversity that existed in Italy, which was where most COVID-positive individuals were returning from. The researchers concluded that “WGS in combination with epidemiological data strengthened the evidence base for public health decision-making in the Netherlands as it enabled a more precise understanding of the transmission patterns in various initial phases of the outbreaks. As such, we were able to understand the genetic diversity of the multiple introduction events in phase 1, the extent of local and regional clusters in phase 2 and the transmission patterns within the HCW [healthcare worker] groups in phase 3 (among which the absence or occurrence of very limited nosocomial transmission)” (Oude Munnink et al., 2020). In Vietnam, a similar application was made regarding a previously known strain responsible for a virus outbreak in the northern region. By WGS, researchers were able to identify the first case of the B.1.1.7 variant from locally acquired infection. As the outbreak expanded, whole genome sequencing enabled enhanced surveillance in high risk groups, like those working in airports, who ended up being assigned another variant of A.23.1, as well as contact tracing and testing to detect more cases (Chau et al., 2021). In China, whole genome sequencing in this initial genomic study was able to provide insight towards the genotype-phenotype differences between COVID-19 positive patients. The researchers concluded, “Pedigree analysis suggested a potential monogenic effect of loss of function variants in GOLGA3 and DPP7 for critically ill and asymptomatic disease demonstration. Genome-wide association study suggests the most significant gene locus associated with severity were located in TMEM189–UBE2V1 that involved in the IL-1 signaling pathway…We identified that the HLA-A*11:01, B*51:01, and C*14:02 alleles significantly predispose the worst outcome of the patients” (F. Wang et al., 2020).
In the United States, a Morbidity and Mortality Weekly Report (MMWR) released in September 2020 utilized serial testing and virus whole genome sequencing at two skilled nursing facilities with COVID-19 outbreaks from April to June 2020 in Minnesota. From a total of 25 specimens from residents at the two different facilities, “strains from 17 residents and five HCP [health care personnel] were genetically similar, including one collected from a dietary worker with limited resident contact. Specimens from two HCP and one resident at facility A had distinctly different virus sequences from the first cluster and from each other. At facility B, 75 (66%) resident specimens and five (7%) HCP specimens were sequenced, all of which were genetically similar”, which suggested “intrafacility transmission”. However, the limited participation by HCPs in serial testing could have “have biased identification of infections and limited interpretation of genomic sequencing” and limited “the description of genetic diversity” (Taylor et al., 2020). Generally, whole genome sequencing still seems to have some limitations, in that “it still presents practical difficulties such as high cost, shortage of available reagents in the global market, need of a specialized laboratorial infrastructure and well-trained staff” resulting in “SARS-CoV-2 surveillance blackouts across several countries” (Bezerra et al., 2021). As of May 4, 2022, there are no FDA approved tests specifically for WGS.
Other types of specimens or media have been proposed as viable for COVID-19 testing, such as saliva. Saliva’s primary advantages include its flexibility, its safety, and overall ease of use in testing. Sri Santosh et al. (2020) also noted that To et al. (2019) found that saliva has a “high consistency rate of greater than 90% with nasopharyngeal specimens in the detection of respiratory viruses, including coronaviruses” (Sri Santosh et al., 2020; To et al., 2019). On Aug. 15, 2020, the FDA issued an EUA to Yale School of Public Health for “SalivaDirect” which uses saliva samples for COVID-19 testing. Although this test still uses RT-PCR, the test still detects the nucleic acids in saliva, but does not require otherwise specialized or proprietary equipment for extraction of those nucleic acids. In the “Performance Evaluation” section of the official EUA, the FDA noted a positive agreement level between SalivaDirect and the ThermoFisher Scientific TaqPath COVID-19 combo kit to be 94.1% (32/34) and a negative agreement level to be 90.9% (30/33) (FDA, 2024a).
A third innovation in COVID-19 testing was published by the FDA on July 18, 2020. On this date, the FDA stated that they reissued an EUA to Quest Diagnostics to authorize Quest SARS-CoV-2 rRT-PCR test for use with “pooled” samples. This testing practice refers to testing multiple samples simultaneously, thereby allowing more efficient testing. The Quest SARS-CoV-2 rRT-PCR test was authorized to test up to four samples at once. The FDA notes that this strategy is most efficient in areas with low prevalence of COVID (i.e., most tests are expected to be negative). In the EUA, the FDA writes that if the “positivity rate” for any given individual to be tested is over 25%, the pooling strategy should not be used due to inefficiency (FDA, 2020). Yelin et al. (2020) found that a single positive sample could be identified in pools of up to 32 samples (with a false negative rate of 10%) and noted that detection of a single positive sample in a pool of 64 samples may be possible with additional amplification cycles (Yelin et al., 2020). Additional EUAs have been issued specifically for tests using pooled samples, such as the UCSD RC SARS-CoV-2 Assay (University of California San Diego Health, RT-PCR, five samples), the Poplar SARS-CoV-2 TMA Pooling assay (Poplar Healthcare, TMA [transcription-mediated amplification], seven samples), and the “COVID-19 RT-PCR Test” (LabCorp, RT-PCR, five samples) (LabCorp, 2022a; Poplar, 2020; UCSD, 2020).
Hogan et al. (2020) performed an analysis of pooled sample analysis in a community setting. The authors analyzed samples in pools of nine or 10, and the RT-PCR assay targeted the envelope (E) gene. When a positive pool was identified, each sample was tested individually for both the E gene and the RNA-dependent RNA polymerase (RdRp) gene for confirmation. The authors investigated 292 pools encompassing 2740 nasopharyngeal samples and 148 bronchoalveolar lavage samples. Two positive samples were identified (0.07%), which both showed detection of both genes. The authors identified one pool with a “positive E signal” that was not reproducible with testing individual samples of that pool. The authors did acknowledge that this methodology may miss individuals in which a COVID-19 risk has not been identified, but concluded that “strategies such as pooled screening may facilitate detection of early community transmission of SARS-CoV-2 and enable timely implementation of appropriate infection control measures to reduce spread (Hogan et al., 2020).
World Health Organization (WHO)
The World Health Organization (WHO) published an interim guideline for the diagnostic testing of “2019 novel coronavirus [termed 2019-nCoV]” on September 11, 2020 (WHO, 2020a). First, they state that routine confirmation of COVID-19 cases is based on nucleic acid testing. Regarding serum testing, they remark that “if negative NAAT results are obtained from a patient in whom SARS-CoV-2 infection is strongly suspected, a paired serum specimen could be collected. One specimen taken in the acute phase and one in the convalescent phase 2 – 4 weeks later can be used to look for seroconversion or a rise in antibody titres.” Finally, they recommend against viral culture or isolation as a routine diagnostic procedure and WHO does not recommend the use of saliva as the sole specimen type for routine clinical diagnostics (WHO, 2020a).
The WHO released a scientific brief with recommendations for the use of SARS-CoV-2 Ag-RDTs and updated their interim guidance on Oct. 6, 2021. Within the guidelines, “SARS-CoV-2 Ag-RDTs (antigen detecting rapid diagnostic tests) that meet the minimum performance requirements of ≥80% sensitivity and ≥ 97% specificity compared to a NAAT reference assay can be used to diagnose SARS-CoV-2 in suspected COVID-19 cases” (WHO, 2021a). Ag-RDTs should be conducted within five to seven days after the onset of symptoms, as “patients who present more than 5-7 days after the onset of symptoms are more likely to have lower viral loads, and the likelihood of false negative results with Ag-RDTs is higher.” WHO recommends that Ag-RDTs be used in settings when they are most reliable — in areas “when there is ongoing community transmission (≥5% test positivity rate). When there is no transmission or low transmission, the positive predictive value of Ag-RDTs will be low (many false positives), and in this setting NAAT is preferable as the first-line testing method or for confirmation of positive Ag-RDTs” (WHO, 2021a).
The WHO recommends using SARS-CoV-2 Ag-RDTs when:
- “Symptomatic individuals (suspected COVID-19 cases) in the first 5-7 days since onset of symptoms”
- For asymptomatic individuals, only “limited to contacts of confirmed or probable cases and to at-risk health workers until more evidence is available on the benefits and cost effectiveness of testing low-risk groups with no known exposure to SARS-CoV-2, particularly in settings where testing capacity is limited.”
- “Suspected COVID-19 cases in outbreak investigations” (WHO, 2021a).
The WHO indicates the following as priority uses for the Ag-RDTs:
- “Community testing of symptomatic individuals meeting the case definition of suspected COVID-19.”
- “To detect and respond to suspected outbreaks of COVID-19 including in remote settings, institutions and semi-closed communities (e.g., schools, care-homes, cruise ships, prisons, workplaces and dormitories), especially where NAAT is not immediately available.”
- “To screen asymptomatic individuals at high risk of COVID-19, including health workers, contacts of cases and other at-risk individuals” (WHO, 2021a).
Overall, “Ag-RDT testing is recommended in settings likely to have the most impact on early detection of cases for care and contact tracing and where test results are most likely to be correct” (WHO, 2021a).
The WHO released a second scientific brief with recommendations concerning immunity passports (WHO, 2020b) on April 24, 2020. Within the guidelines, WHO states that as of the publication date, “no study has evaluated whether the presence of antibodies to SARS-CoV-2 confers immunity to subsequent infection by this virus in humans.” They go on to note, “Laboratory tests that detect antibodies to SARS-CoV-2 in people, including rapid immunodiagnostic tests, need further validation to determine their accuracy and reliability. Inaccurate immunodiagnostic tests may falsely categorize people in two ways. The first is that they may falsely label people who have been infected as negative, and the second is that people who have not been infected are falsely labelled as positive. Both errors have serious consequences and will affect control efforts. These tests also need to accurately distinguish between past infections from SARS-CoV-2 and those caused by the known set of six human coronaviruses. Four of these viruses cause the common cold and circulate widely. The remaining two are the viruses that cause Middle East Respiratory Syndrome and Severe Acute Respiratory Syndrome. People infected by any one of these viruses may produce antibodies that cross-react with antibodies produced in response to infection with SARS-CoV-2” (WHO, 2020b).
In 2021, WHO released an update to the scientific brief concerning immunity passports within a document titled ‘COVID-19 natural immunity.’ Within this brief, WHO discusses the various testing methods available. WHO notes that “there are many available serologic assays that measure the antibody response to SARS-CoV-2 infection, but at the present time, the correlates of protection are not well understood.” The most measured immune response is the presence of antibodies in serum. Serologic assays to detect the antibody response are usually based on enzyme immunoassays, which detect the presence of virus-specific antibodies in the blood or by live or pseudo-virus neutralization assays, which detect functional NAb. While serologic testing has limited use in clinical management because it does not capture active infection, it can be very useful in determining the extent of infection or estimating attack rates in given populations. Interpreting the results of serologic testing, however, is complex: there are several antibody types and subtypes and multiple antigenic determinants/epitopes that can be used to target these antibodies, and the results may differ substantially depending on the combinations chosen. The results will also depend on the manufacturing specifics of the assay used”. Other frequently used assays are enzyme-linked immunosorbent tests, chemiluminescent tests, and lateral flow rapid diagnostic tests. To conclude, “available tests and current knowledge do not tell us about the duration of immunity and protection against reinfection, but recent evidence suggests that natural infection may provide similar protection against symptomatic disease as vaccination, at least for the available follow up period” (WHO, 2021c).
The WHO released guidelines for the use of SARS-CoV-2 antigen-detection rapid diagnostic tests for COVID-19 self-testing. The key points are:
- “COVID-19 self-testing, using SARS-CoV-2 Ag-RDTs, should be offered in addition to professionally administered testing services (Strong recommendation, low to moderate certainty evidence). This recommendation is based on evidence that shows users can reliably and accurately self-test, and that COVID-19 self-testing is acceptable and feasible and may reduce existing inequalities in testing access.
- The role and use of COVID-19 self-testing — including why, where and how it should be used — will need to be adapted to national priorities, epidemiology, resource availability, and local context with community input. Clear and up-to-date messaging will be needed to ensure self-test users can understand when to test, the meaning of their test results and post-test responsibilities.
- Self-testing should always be voluntary and never mandatory or coercive. It is important that in certain settings, such as schools and workplaces, self-testing costs are not borne by students or workers.
- Access to affordable and quality-assured SARS-CoV-2 Ag-RDTs, including for self-testing, should particularly be prioritized for settings where there is limited access to NAAT. COVID-19 self-test kits should meet the existing World Health Organization (WHO) standards for Ag-RDTs (≥ 80% sensitivity and ≥ 97% specificity among symptomatic individuals).
- COVID-19 self-testing can be considered for both diagnostic and screening purposes. Depending on the epidemiological situation, a positive self-test result in symptomatic individuals or those with recent exposure could be used for diagnosis, and to facilitate linkage to clinical care and therapeutics.
- For screening purposes, a negative self-test result could enable participation in an activity, such as group activities or indoor gatherings, and confirmatory testing for positive results can be considered.
- Each country is facing a different situation in the pandemic depending on several factors including the intensity of SARS-CoV-2 circulation, amount of population level immunity, capacities to respond and agility to adjust measures. Timely and accurate diagnostic testing for SARS-CoV-2, the virus that causes COVID-19, is an essential part of a comprehensive COVID-19 response strategy. As the pandemic continues and the virus evolves, policy adjustments related to SARS-CoV-2 testing approaches and services, including COVID-19 self-testing, will be needed” (WHO, 2022).
The WHO released a scientific brief on May 15, 2020, concerning multisystem inflammatory syndrome in children and adolescents with COVID-19. Within the guidelines, they recommend standardized data describing clinical presentations.
- The WHO gives a preliminary case definition for individuals ages 0 – 19 years with fever three or more days AND at least TWO of the following:
- “Rash or bilateral non-purulent conjunctivitis or muco-cutaneous inflammation signs (oral, hands or feet).
- Hypotension or shock.
- Features of myocardial dysfunction, pericarditis, valvulitis, or coronary abnormalities (including [echocardiogram] findings or elevated Troponin/NT-proBNP).
- Evidence of coagulopathy (by PT, PTT, elevated d-Dimers).
- Acute gastrointestinal problems (diarrhea, vomiting, or abdominal pain).
- AND
- Elevated markers of inflammation such as ESR, C-reactive protein, or procalcitonin.
- AND
- No other obvious microbial cause of inflammation, including bacterial sepsis, staphylococcal or streptococcal shock syndromes.
- AND
- Evidence of COVID-19 (RT-PCR, antigen test or serology positive), or likely contact with patients with COVID-19” (WHO, 2020c).
Centers for Disease Control and Prevention (CDC)
In the CDC guidelines, Testing for COVID-19, there are two main types of viral tests used to detect current infections of SARS-CoV-2. Nucleic acid amplification tests (NAATs), which includes PCR tests, are the most highly recommended as they are highly sensitive and highly specific tests that detect one or more viral ribonucleic acid (RNA) genes. Viral RNA may stay in a person's body for up to 90 days after they test positive. Therefore, NAATs should not be used to test someone who has tested positive in the last 90 days (CDC, 2024f, 2024h).
Antigen tests are rapid tests that can produce results in 15-30 minutes. They are immunoassays that detect the presence of specific viral proteins, called antigens. Antigen tests generally have high specificity, similar to NAATs, but are less sensitive than most NAATs. Therefore, “positive results are accurate and reliable. However, in general, antigen tests are less likely to detect the virus than NAAT tests, especially when symptoms are not present. Therefore, a single negative antigen test cannot rule out infection.” The CDC recommends two negative antigen tests for individuals with symptoms or three antigen tests for those without symptoms, performed 48 hours apart to confirm an individual does not have COVID-19. However, a single NAAT test can be used to confirm an antigen test result (CDC, 2024f, 2024h).
- If an individual has not had COVID-19 or has not had a positive test within the past 90 days: they may choose a NAAT, including PCR, or antigen test. If the antigen test result is negative, repeat testing following the recommendations above.
- If an individual has tested positive for COVID-19 within the past 30 days or less with symptoms: use an antigen test. Repeat negative tests following the recommendations above.
- If an individual has tested positive for COVID-19 within the past 30 days or less with no symptoms: testing is not recommended to detect a new infection.
- If an individual has tested positive for COVID-19 within the 31-90 days with or without symptoms: use an antigen test. Repeat negative tests following the recommendations above (CDC, 2024h).
After a positive test result, you may continue to test positive for some time. Some tests, especially NAAT tests, may continue to show a positive result for up to 90 days. Reinfections can occur within 90 days, which can make it hard to know if a positive test indicates a new infection. Consider consulting a healthcare provider if you have any questions or concerns about your circumstances (CDC, 2024h).
Antibody (or serology) tests are used to test for the presence of antibodies from previous infection or vaccination and can be used in the diagnosis of Multisystem Inflammatory Syndrome in Children (MIS-C) or Multisystem Inflammatory Syndrome in Adults (MIS-A). However, antibody testing does not diagnose current infection. Antibody testing is not currently recommended to assess a person's protection against SARS-CoV-2 infection or severe COVID-19 following COVID-19 vaccination or prior infection, or to assess the need for vaccination in an unvaccinated person (CDC, 2024f).
In the CDC guidelines, MIS Case Definitions and Reporting, they define cases for MIS-C and MIS-A associated with SARS-CoV-2 infection. MIS is a rare but serious condition associated with SARS-CoV-2, in which different body parts become inflamed such as heart, lungs kidneys, brain, skin, eyes, and gastrointestinal tract. Children and adults with MIS experience ongoing fever PLUS more than one of the following: stomach pain, bloodshot eyes, diarrhea, dizziness or lightheadedness (signs of low blood pressure), skin rash, vomiting (CDC, 2024e). MIS-C is defined as any illness in a person < 21 years of age that meets:
- “The clinical AND the laboratory criteria (Confirmed); OR
- The clinical criteria AND epidemiologic linkage criteria (Probable); OR
- The vital records criteria (Suspect)”
Clinical Criteria: An illness characterized by all of the following, in the absence of a more likely alternative diagnosis*
- “Subjective or documented fever (temperature ≥ 38.0⁰ C)
- Clinical severity requiring hospitalization or resulting in death
- Evidence of systemic inflammation indicated by C-reactive protein ≥ 3.0 mg/dL (30 mg/L)
- New onset manifestations in at least two of the following categories:
- Cardiac involvement indicated by: Left ventricular ejection fraction < 55% OR coronary artery dilatation, aneurysm, or ectasia, OR troponin elevated above laboratory normal range, or indicated as elevated in a clinical note
- Mucocutaneous involvement indicated by: Rash, OR inflammation of the oral mucosa (e.g., mucosal erythema or swelling, drying or fissuring of the lips, strawberry tongue), OR conjunctivitis or conjunctival injection (redness of the eyes), OR extremity findings (e.g., erythema [redness] or edema [swelling] of the hands or feet)
- Shock**
- Gastrointestinal involvement indicated by: Abdominal pain, OR Vomiting, OR Diarrhea
- Hematologic involvement indicated by: Platelet count <150,000 cells/uL, OR absolute lymphocyte count (ALC)”
Laboratory Criteria:
- “Detection of SARS-CoV-2 RNA in a clinical specimen*** up to 60 days prior to or during hospitalization, or in a post-mortem specimen using a diagnostic molecular amplification test (e.g., polymerase chain reaction [PCR]), OR
- Detection of SARS-CoV-2 specific antigen in a clinical specimen*** up to 60 days prior to or during hospitalization, or in a post-mortem specimen, OR
- Detection of SARS-CoV-2 specific antibodies^ in serum, plasma, or whole blood associated with current illness resulting in or during hospitalization”
Epidemiological Linkage Criteria: “Close contact‡ with a confirmed or probable case of COVID-19 disease in the 60 days prior to hospitalization.”
Vital Records Criteria: “A person whose death certificate lists MIS-C or multisystem inflammatory syndrome as an underlying cause of death or a significant condition contributing to death”
“*If documented by the clinical treatment team, a final diagnosis of Kawasaki Disease should be considered an alternative diagnosis. These cases should not be reported to national MIS-C surveillance.
**Clinician documentation of shock meets this criterion.
***Positive molecular or antigen results from self-administered testing using over-the-counter test kits meet laboratory criteria.
">^Includes a positive serology test regardless of COVID-19 vaccination status. Detection of anti-nucleocapsid antibody is indicative of SARS-CoV-2 infection, while anti-spike protein antibody may be induced either by COVID-19 vaccination or by SARS-CoV-2 infection
‡Close contact is generally defined as being within 6 feet for at least 15 minutes (cumulative over a 24-hour period). However, it depends on the exposure level and setting; for example, in the setting of an aerosol generating procedure in healthcare settings without proper personal protective equipment (PPE), this may be defined as any duration” (CDC, 2024e).
The CDC defines MIS-A as an illness in a person ≥ 21 years of age with:
- “Hospitalization for ≥ 24 hours* AND
- Subjective of documented fever (≥ 38.0 C) for ≥ 24 hours prior to hospitalization or within the first THREE days of hospitalization AND
- An illness meeting the following clinical and laboratory criteria:”
Clinical Criteria: “No alternative diagnosis (e.g. bacterial sepsis, exacerbation of a chronic medical condition) AND at least THREE of the following clinical criteria occurring prior to hospitalization or within the first THREE days of hospitalization. At least ONE must be a primary clinical criterion.
- Primary clinical criteria: Severe cardiac illness** (Includes myocarditis, pericarditis, coronary artery dilatation/aneurysm, new-onset right or left ventricular dysfunction (LVEF< 50%), 2nd/3rd degree A-V block, or ventricular tachycardia). Rash AND non-purulent conjunctivitis
- Secondary clinical criteria: New-onset neurologic signs and symptoms (Includes encephalopathy in a patient without prior cognitive impairment, seizures, meningeal signs, or peripheral neuropathy including Guillain-Barré syndrome). Shock or hypotension not attributable to medical therapy (e.g., sedation, renal replacement therapy). Abdominal pain, vomiting, or diarrhea. Thrombocytopenia (platelet count <150,000/ microliter. “
Laboratory Criteria: “Evidence of SARS-CoV-2 infection (positive SARS-CoV-2 nucleic acid amplification (NAAT), serology, or antigen test) AND evidence of systemic inflammation (elevated levels of at least 2 of the following: C-reactive protein (CRP), ferritin, interleukin-6 (IL-6), erythrocyte sedimentation rate (ESR), procalcitonin). “
“*Or hospitalized for any length of time with an illness resulting in death
**Cardiac arrest alone does not meet this criterion” (CDC, 2024e).
According to the CDC, long COVID, also known as post-COVID conditions (PCC) is “an infection-associated chronic condition that can occur after SARS-CoV-2 infection and is present for at least 3 months as a continuous, relapsing and remitting, or progressive disease state that affects one or more organ systems” (CDC, 2024d). Long COVID is associated with:
- “Development of new or recurrent symptoms and conditions after the symptoms of initial acute COVID-19 illness have resolved.
- Symptoms that can emerge, persist, resolve, and reemerge over varying lengths of time.
- A spectrum of physical, social, and psychological consequences.
- Functional limitations that can affect patient wellness and quality of life”
Clinicians may clinically evaluate and diagnose Long COVID based on patient history and findings from a physical examination, while others might require directed diagnostic testing. Currently, no laboratory test can be used to definitively diagnose Long COVID or to distinguish Long COVID from conditions with different etiologies. Objective laboratory or imaging findings should not be used as the only measure or assessment of a patient's well-being. For example, a positive SARS-CoV-2 viral test or serologic (antibody) test are not required to establish a diagnosis of Long COVID but can help assess for current or previous infection.
A wide range of symptoms and clinical findings can occur in people with varying degrees of illness from acute SARS-CoV-2 infection. These effects can overlap with multiorgan complications, or with effects of treatment or hospitalization and can persist after the acute COVID-19 illness has resolved. While more than 200 Long COVID symptoms have been identified, commonly reported symptoms include:
- “Bloating/constipation/diarrhea
- Difficulty concentrating
- Light headedness/fast heart rate
- Memory change
- Persistent fatigue
- Post-exertional malaise
- Problems with smell
- Problems with taste
- Recurring headaches
- Shortness of breath/cough
- Sleep disturbance” (CDC, 2024d).
Post-exertional malaise (PEM) is the worsening of symptoms following even minor physical or mental exertion, with symptoms typically worsening 12 to 48 hours after activity and lasting for days or even weeks.
National Institutes of Health (NIH)
The NIH updated their COVID-19 treatment guidelines in May of 2024. The NIH addresses the clinical spectrum of SARS-CoV-2 infection, which includes those with asymptomatic or presymptomatic infection, mild illness, moderate illness, severe illness, and critical illness. For asymptomatic and presymptomatic individuals, the NIH states that “the percentage of individuals who present with asymptomatic infection and progress to clinical disease is unclear. Some asymptomatic individuals have been reported to have objective radiographic findings consistent with COVID-19 pneumonia.” Additionally, the guideline discusses infectious complications in patients with COVID-19, which can be categorized as “coinfections at presentation,” such as “concomitant viral infections, including influenza and other respiratory viruses” and community-acquired bacterial pneumonia, and “reactivation of latent infections,” such as chronic hepatitis B virus and latent tuberculosis reactivation, “nosocomial infections,” such as hospital-acquired or ventilator-associated pneumonia and Clostridioides difficile-associated diarrhea, and “opportunistic fungal infections,” like aspergillosis and mucormycosis among hospitalized COVID-19 patients (NIH, 2024a).
The NIH also released COVID-19 testing guidelines. The following recommendations were made from the COVID-19 Treatment Guidelines Panel:
- The Panel recommends “using either a nucleic acid amplification test (NAAT) or an antigen test with a sample collected from the upper respiratory tract (e.g., nasopharyngeal, nasal mid-turbinate, or anterior nasal) to diagnose acute infection of SARS-CoV-2 (AIII).”
- “A NAAT should not be repeated in an asymptomatic person (with the exception of health care workers) within 90 days of a previous SARS-CoV-2 infection, even if the person has had a significant exposure to SARS-CoV-2.”
- “SARS-CoV-2 reinfection has been reported in people after an initial diagnosis of the infection; therefore, clinicians should consider using a NAAT for those who have recovered from a previous infection and who present with symptoms that are compatible with SARS-CoV-2 infection if there is no alternative diagnosis (BIII).”
- “The Panel recommends against diagnosing acute SARS-CoV-2 infection solely on the basis of serologic (i.e., antibody) test results (AIII).”
- “There is insufficient evidence for the Panel to recommend either for or against the use of SARS-CoV-2 serologic testing to assess for immunity or to guide clinical decisions about using COVID-19 vaccines or anti-SARS-CoV-2 monoclonal antibodies” (NIH, 2024b).
American Medical Association (AMA)
The AMA released public health guidelines and recommendations concerning serological testing for SARS-CoV-2 antibodies on May 14, 2020. They list the limitations of antibody testing to include the potential for false-positive results, potential cross-reactivity, and lack of knowledge concerning relationship between antibody testing and immune status. The AMA recommends the following:
- “Use of serology tests should currently be limited to population-level seroprevalence study, evaluation of recovered individuals for convalescent plasma donations, and in other situations where they are used as part of a well-defined testing plan and in concert with other clinical information by physicians well-versed in interpretation of serology test results.”
- “Serology tests should not be offered to individuals as a method of determining immune status.”
- “Serology tests should not currently be used as the basis for any “immunity certificates,” to inform decisions to return to work, or to otherwise inform physical distancing decisions. Doing so may put individuals, their household and their community at risk.”
- “Serology tests should not be used as the sole basis of diagnosis of COVID-19 infection” (AMA, 2020).
“Messaging on serological testing to medically underserved communities should explicitly take into consideration cultural and social features which may bear on their ability to make long-term choices on physical distancing and other COVID-19 precautions” (AMA, 2020).
Infectious Diseases Society of America (IDSA)
The IDSA released guidelines on the molecular diagnostic testing for COVID-19 which includes the following recommendations (IDSA, 2023):
“Recommendation 1: The IDSA panel recommends a SARS-CoV-2 NAAT in symptomatic individuals suspected of having COVID-19 (strong recommendation, moderate certainty evidence).
- Remarks:
- The panel considered symptomatic patients to have at least one of the most common symptoms compatible with COVID-19 (Fever or chills, cough, shortness of breath or difficulty breathing, fatigue, muscle or body aches, headache, sore throat, new loss of taste or smell, congestion or runny nose, nausea or vomiting, diarrhea).
- A positive test result may inform decisions about therapy, isolation, and potentially contact tracing.
There were limited data available regarding the analytical performance of SARS-CoV-2 NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, in children, or in patients infected with recent SARS-CoV-2 variants (e.g., Omicron).
Recommendation 2: For symptomatic individuals suspected of having COVID-19, the IDSA panel suggests collecting and testing swab specimens from either the nasopharynx (NP), anterior nares (AN), oropharynx (OP), or midturbinate regions (MT); saliva, or mouth gargle (conditional recommendation, low certainty evidence).
- Remarks:
- Compared to NP swabs, AN or OP swabs alone yield more false-negative results than combined AN/OP swabs, MT swabs, saliva, or mouth gargle. Swabs of AN or OP alone are acceptable if collection of NP, AN/OP, or MT swabs, saliva, or mouth gargle is not feasible.
- Sample collection methods are not standardized (e.g., drool or spit with/without cough were all reported as saliva)
- The patient’s ability to follow instructions and cooperate with requirements of specimen collection (e.g., spit into a container, nothing by mouth for some time before saliva collection) should be considered.
FDA approval of individual NAATs specifically indicates collection and specimen type(s). Failure to adhere to label requirements, unless otherwise approved through a lab developed test (LDT) validation or authorized by the FDA through a subsequent EUA for different collection or specimen type, can lead to inaccurate results and reimbursement denials.
Recommendation 3: The IDSA panel suggests that for symptomatic individuals suspected of having COVID-19, AN and MT swab specimens may be collected for SARS-CoV-2 RNA testing by either patients or healthcare providers (conditional recommendation, moderate certainty evidence).
- Remarks:
- An important limitation of the data available to inform this recommendation is that the type of specimen differed by comparison group. That is, while self-collected samples were always AN and MT specimens, healthcare provider-collected samples were always NP specimens. This might explain the increased sensitivity of healthcare provider collected specimens.
Recommendation 4: The IDSA panel suggests using either rapid or standard laboratory-based NAATs in symptomatic individuals suspected of having COVID-19 (conditional recommendation, moderate certainty of evidence).
- Remarks:
- Appropriate specimen collection and transport to the laboratory or testing site are critical to ensuring high-quality results; resources are available on the IDSA website. Definitions of rapid NAATs have varied; some, including the U.S. FDA, consider turnaround times less than or equal to 30 minutes to define rapid NAATs, whereas others use less than or equal to 60-minutes or even longer. This time is for testing only (inclusive of nucleic acid extraction) and does not include time between specimen collection and testing or time between testing and reporting. Rapid tests typically have few operator steps and may be amendable to testing near patients or even at the point-of-care performed by non-laboratory staff. Rapid molecular test methodologies include rapid reverse transcription polymerase chain reaction (RT-PCR) and rapid isothermal NAAT. Standard tests require instrumentation and/or processing that must typically be performed in a clinical laboratory by trained laboratory staff.
This recommendation applies only to tests evaluated in the included studies. One test, Abbott IDNow, was included in most of the studies evaluated in this recommendation and may have skewed results towards lower sensitivity. Variability of test performance with different specimen types may be important. The evaluated assays used diverse technologies (e.g., isothermal and non-isothermal test amplification) that may theoretically impact results. Limited data were available regarding the analytical performance of NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, or in those infected with contemporary SARS-CoV-2 variants.
Recommendation 5: The IDSA panel suggests performing a single NAAT and not repeating testing routinely in symptomatic or asymptomatic individuals suspected of having COVID-19 whose initial NAAT result is negative (conditional recommendation, very low certainty of evidence).
- Remarks:
- The panel considered symptomatic patients to have at least one of the most common symptoms compatible with COVID-19
- While repeat testing when the initial test result is negative is not suggested routinely, there may be situations where repeat testing might be considered. An example of such a situation is the development of new or worsening symptoms compatible with COVID-19 in the absence of an alternative explanation. Also, timing of symptom onset might drive a need for repeat testing. A poorly collected specimen could yield a falsely negative result and might be another reason for repeat testing.
If performed, repeat testing should generally occur 24 – 48 hours after initial testing and once the initial NAAT result has returned as negative.
Recommendation 6: For individuals who have clinical or epidemiologic reasons that might make testing desirable, the IDSA panel suggests SARS-CoV-2 RNA testing in asymptomatic individuals who are either known or suspected to have been exposed to COVID-19 (conditional recommendation, moderate certainty evidence).
- Remarks:
- The panel recognizes the lack of evidence supporting therapy for asymptomatic persons and the absence of treatment approved through EUA for asymptomatic COVID-19, but acknowledges that individual clinical scenarios may lead clinicians toward testing and consideration of treatment. Individuals who have clinical or epidemiologic reasons that might make testing desirable (e.g., high-risk individuals, such as those who have pulmonary conditions or are immunocompromised or those in close contact with immunocompromised individuals) may be considered for testing. Testing should be done at least 5 days after the exposure. If symptoms develop before 5 days, the exposed individual should get tested immediately[3]. Knowledge that an individual is infected with SARS-CoV-2 can be helpful to inform appropriate isolation. The decision to test asymptomatic persons should depend on the availability of testing resources. Known exposures are defined herein as close contact for at least 15 minutes over a 24-hour period with someone who has laboratory-confirmed COVID-19. Suspected exposures might be defined as working or residing in a congregate setting (e.g., long-term care or correctional facility, cruise ship, factory) experiencing a COVID-19 outbreak. The risk of contracting SARS-CoV-2 may vary under different exposure conditions, e.g., length of time exposed, indoor versus outdoor setting, whether masks were routinely worn. Household contacts may be especially high-risk. This recommendation assumes the exposed individual was not wearing appropriate PPE.
Recommendation 7: For individuals who have clinical or epidemiologic reasons that might make testing desirable, the IDSA panel suggests using either rapid or laboratory-based NAATs in asymptomatic individuals with known exposure to SARS-CoV-2 infection (conditional recommendation, moderate certainty of evidence).
- Remarks:
- Appropriate specimen collection and transport to the laboratory or testing site are critical to ensure quality results; resources are available on the IDSA website. Definitions of rapid NAATs have varied; some, including the U.S. FDA, consider turnaround times less than or equal to 30 minutes to define rapid NAATs, whereas others use less than or equal to 60-minutes or even longer. This time is for testing only (inclusive of nucleic acid extraction) and does not include time between specimen collection and testing or time between testing and reporting. Rapid tests typically have few operator steps and may be amendable to testing near patients or even at the point-of-care performed by non-laboratory staff. Rapid test methodologies include rapid RT-PCR and rapid isothermal NAAT. Standard tests require instrumentation and/or processing that must typically be performed in a clinical laboratory by trained laboratory staff.
This recommendation applies only to tests evaluated in the included studies. Variability of test performance with different specimen types may be important. The evaluated assays used diverse technologies (e.g., isothermal and non-isothermal test amplification) that may theoretically impact results. Limited data were available regarding the analytical performance of NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, or in those infected with different SARS-CoV-2 variants.
Recommendation 8: The IDSA panel suggests against routine SARS-CoV-2 NAAT in asymptomatic individuals without a known exposure to COVID-19 who are being hospitalized (conditional recommendation, very low certainty evidence).
- Remarks:
- Important considerations for this recommendation are that the IDSA panel was unable to identify studies published during the period of literature review that showed reduced SARS-CoV-2 transmission to healthcare providers or to other patients resulting from prehospitalization testing. The evidence was indirect and assessed only diagnostic test accuracy in studies of symptomatic patients alone or together with asymptomatic patients. The burden of testing all patients planned to be admitted was considered, in the face of limited evidence. Finally, there are other effective infection prevention interventions, including use of PPE and vaccination that should be considered.
The panel acknowledges that there could be a benefit of pre-admission NAAT in some situations, such as admission to a multibed room; to a unit with a congregate treatment area, such as a behavioral health unit; or to a positive pressure room or unit.
Recommendation 9: The IDSA panel suggests against routine SARS-CoV-2 NAAT of asymptomatic individuals without a known exposure to COVID-19 who are undergoing a medical or surgical procedure (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens without evidence of infectious virus has been reported widely.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on non-standardized instrument signal values, such as cycle threshold (Ct) values.
- Decisions on the timing of infection must balance the risk to the patient against the risks of delaying or avoiding the planned procedure, and should consider patient-related factors (e.g., vaccination status, symptomatic status, age), procedure-related factors (e.g., level of urgency, whether procedure generates aerosols), and procedural area infection control practices.
- Given limited evidence for poor outcomes in asymptomatic persons who undergo major surgery soon after testing positive for SARS-CoV-2 infection, testing may be considered during periods of high community transmission.
- Testing may also be considered before solid organ transplantation, hematopoietic stem cell transplantation or CAR-T cell therapy.
This recommendation applies to settings where protective measures, such as PPE, are available and are used with adherence. Other factors to consider include the vaccination status of healthcare providers and patients, and whether patients will be roomed with other patients before or after the procedure. This recommendation is based on general exposure in the community as compared to a specific known exposure.
Recommendation 10: The IDSA panel suggests against routinely repeating NAAT before medical or surgical procedures in patients with a recent history of COVID-19 (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens without evidence of infectious virus has been reported widely.
- Conversely, the IDSA panel was unable to find definitive evidence demonstrating that a negative NAAT result following a positive result is proof that a patient is no longer infectious.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on Ct value results.
Decisions on the timing of a procedure in a patient with prior SARS-CoV-2 infection must balance the risk to the patient against the risks of delaying or avoiding the planned procedure, and should consider patient-related factors (e.g., vaccination status, symptomatic status, age), procedure-related factors (e.g., level of urgency, whether procedure generates aerosols), and procedural area infection control practices.
Recommendation 11: The IDSA panel suggests against routinely repeating NAAT in patients with COVID-19 to guide release from isolation (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens for prolonged periods without evidence of infectious virus has been reported widely. Predicating release from isolation on a negative SARS-CoV-2 NAAT may extend the duration of isolation unnecessarily.
- Conversely, the IDSA panel was unable to find definitive evidence demonstrating that a negative NAAT result following a positive result is proof that a patient is no longer infectious.
The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on Ct value results.
Recommendation 12: The IDSA panel suggests neither for nor against home-testing for SARS-CoV-2. (evidence gap).
- Remarks:
- The panel defined time-sensitive surgery as medically necessary surgeries that need to be done within three months.
- Testing should ideally be performed as close to the planned surgery as possible (e.g., within 48 – 72 hours).
- To limit potential poor outcomes, deferring non-emergent surgeries should be considered for patients testing positive for SARS-CoV-2.
- Decisions about PPE use for the aerosol generating portions of these procedures may be dependent on test results when there is limited availability of PPE. However, there is a risk for false negative test results, so caution should be exercised by those who will be in close contact with/exposed to the upper respiratory tract (e.g., anesthesia personnel, ENT procedures).
The decision to test asymptomatic patients will be dependent on the availability of testing resources. This recommendation does not address the need for repeat testing if patients are required to undergo multiple surgeries over time” (IDSA, 2023).
In total, the IDSA panel made 12 recommendations for SARS-CoV-2 nucleic acid testing based on new systematic reviews of the diagnostic literature. An updated algorithm based on these recommendations is provided to aid in decision-making seen below (IDSA, 2023).
The IDSA also published a guideline regarding serology testing with the following recommendations (IDSA, 2024):
- “The IDSA panel recommends against using serologic testing to diagnose SARS-CoV-2 infection during the first two weeks following symptom onset (strong recommendation, low certainty of evidence).
- The IDSA panel recommends against using IgG antibodies to provide evidence of COVID-19 in symptomatic patients with a high clinical suspicion and repeatedly negative NAAT (strong recommendation, very low certainty of evidence).
- To assist with the diagnosis of multisystem inflammatory syndrome in children (MIS-C), the IDSA panel recommends using both IgG antibody testing and NAAT to provide evidence of current or recent past COVID-19 (strong recommendation, very low certainty of evidence).
- When evidence of previous SARS-CoV-2 infection is desired, the IDSA panel suggests testing for SARS-CoV-2 IgG, IgG/IgM, or total antibodies three to five weeks after symptom onset and suggests against testing for SARS-CoV-2 IgM (conditional recommendation, low certainty of evidence).
- When evidence of prior SARS-CoV-2 infection is desired, the IDSA panel suggests using serologic assays that target nucleocapsid protein rather than spike protein (conditional recommendation, low certainty of evidence).
- In individuals with previous SARS-CoV-2 infection or vaccination, the IDSA panel suggests against routine serologic testing given no demonstrated benefit to improving patient outcomes (conditional recommendation, very low certainty of evidence)” (IDSA, 2024).
Infectious Diseases Society of America (IDSA)/American Society for Microbiology (ASM)
In 2022, IDSA and ASM released a consensus review document on the clinical and infection prevention applications for SARS-CoV-2 genotyping. In it, they cover clinical use cases for genotyping, methods of genotyping, assay validation and regulatory requirements, clinical reporting for laboratories, and emerging issues in clinical SARS-CoV-2 sequencing. Overall, they report that “while clinical uses of SARS-CoV2 genotyping are currently limited, rapid technological change along with a growing ability to interpret variants in real time foretell a growing role for SARS-CoV-2 genotyping in clinical care as continuing data emerge on vaccine and therapeutic efficacy” (Greninger et al., 2022).
Society for Healthcare Epidemiology of America- (SHEA)/American Society of Anesthesiologists (ASA)/Anesthesia Patient Safety Foundation (APSF)
In late 2022, SHEA published recommendations on screening for SARS-CoV-2 in an asymptomatic population. Here, they note that testing of asymptomatic patients was an attempt to reduce the risk of nosocomial transmission but has been an extensive and resource intensive process with unclear benefit when added to other layers of infection prevention mitigation controls. They also note that “the logistic challenges and costs related to screening program implementation, data noting the lack of substantial aerosol generation with elective controlled intubation, extubation, and other procedures, and the adverse patient and facility consequences of asymptomatic screening call into question the utility of this infection prevention intervention.” Based on their findings, SHEA “recommends against routine universal use of asymptomatic screening for SARS-CoV-2 in healthcare facilities. Specifically, preprocedure asymptomatic screening is unlikely to provide incremental benefit in preventing SARS-CoV-2 transmission in the procedural and perioperative environment when other infection prevention strategies are in place, and it should not be considered a requirement for all patients. Admission screening may be beneficial during times of increased virus transmission in some settings where other layers of controls are limited (eg, behavioral health, congregate care, or shared patient rooms), but widespread routine use of admission asymptomatic screening is not recommended over strengthening other infection prevention controls” (Talbot et al., 2023).
This statement is supported by the ASA and the APSF. They specifically note that the “SHEA recommendations provide a rationale for considering a move away from universal screening. Such a change considers the potential adverse consequences of testing for SARS-CoV-2 in asymptomatic patients. Moreover, we recommend that each facility develop a risk/benefit analysis that includes local/facility infection prevention assessment (e.g., patient population, facility physical layout, and community incidence and transmission of COVID-19 as defined in the SHEA Board Commentary), and a robust system of controls and interventions to prevent virus transmission (“Swiss Cheese” model). The recommendations by SHEA should be considered along with these updated recommendations to operationalize a robust and safe perioperative screening and targeted testing program for the benefit of our patients, our healthcare workers, other hospital patients and the public” (ASA & APSF, 2022).
American Association for Clinical Chemistry (AACC)
The AACC released a set of recommendations for “implementing and interpreting SARS-CoV-2 EUA and LDT serologic testing in clinical laboratories.” Serologic testing is currently only used for serum, plasma, and “less frequently, whole-blood or dried blood spots,” but not for other sample types, like saliva and cerebrospinal fluid. Serologic testing is “not recommended as the primary approach for diagnosis of SARS-CoV-2 infection.” For the recommended use of serologic testing, the AACC stated the following:
- “Serologic testing may be offered as an approach to support diagnosis of COVID -19 illness in symptomatic patients and late phase negative molecular testing or for patients presenting with late complications such as multisystem inflammatory syndrome in children (MIS -C).
- Serologic testing can help identify people who may have been infected with or have recovered from the SARS -CoV -2 infection.
- Serologic testing can be used to screen potential convalescent plasma donors and in the manufacture of convalescent plasma.
- Serologic testing can be used for epidemiology and seroprevalence studies.
- Serologic testing can be used for vaccine response and efficacy studies.”
Regarding serologic testing limitations, the AACC stated the following:
- “False positive results may occur.
- Negative results do not preclude acute SARS-CoV-2 infection or viral shedding.
- Serologic tests may not differentiate between natural infection and vaccine response.
- Serologic results should not be used for
- Determining individual protective immunity
- Return to work decisions
- Cohorting individuals in congregate settings
- Assessment of convalescent plasma recipients
- Use of Personal Protective Equipment
- Placement of high-risk job functions” (Zhang et al., 2021).
European Centre for Disease Prevention and Control (ECDC)
The ECDC in their guidance for laboratory support in the EU/EEA recommends using WHO-recommended testing strategies for the diagnosis and confirmation of COVID-19 (ECDC, 2023).
In the ECDC’s guideline titled “COVID-19 testing strategies and objectives”, the ECDC recommends performing laboratory testing in accordance with the WHO case definition. The following populations should be tested (ECDC, 2022b):
- “Ideally, all people with COVID-19 symptoms should be tested as soon as possible after symptom onset. This requires easy access to testing for all, including non-residents. Test result turnaround time should be minimized, people testing positive should isolate and timely contact tracing should be carried out, ensuring that all close contacts are tested, irrespective of symptoms.
- All patients with acute respiratory symptoms in hospitals and in other healthcare settings, and all specimens from sentinel primary care surveillance should be tested for both SARS-CoV-2 and influenza during the influenza season to monitor incidence and trends over time.
- Healthcare and social care settings require intensive testing when there is documented community transmission. Periodic and comprehensive testing of all staff and residents/patients is recommended to prevent nosocomial transmission. Furthermore, all patients/residents should be tested upon or just prior to admission.
- Clusters or outbreaks may occur in certain settings, such as workplaces, educational facilities, prisons, and migrant detention centres. Testing policies and systems should be in place for rapid detection and control to protect the relevant populations in these settings and to protect the community from amplified transmission.
- Countries experiencing high SARS-CoV-2 transmission in a local community should consider testing the whole population of the affected area. This would enable identification of infectious COVID-19 cases and allow for their prompt isolation to interrupt chains of transmission. Depending on the epidemiological situation, size and population density of the affected area, such an approach could be less disruptive for society than having to introduce and ensure compliance with more stringent public health measures.
- To prevent re-introduction, countries or subnational areas that achieved sustained control of the circulation of SARS-CoV-2 should, in addition to quarantine measures, consider targeted testing and follow-up of individuals coming from other areas within the same country, or from other countries that have not yet achieved sustained control of the virus” (ECDC, 2022b).
The ECDC notes that “Genomic surveillance of SARS-CoV-2 is essential to detect, monitor and assess virus variants that can result in increased transmissibility, disease severity, or have other adverse effects on public health and social control measures. Obtaining timely and accurate information on the emergence and circulation of variants of concern (VOCs) and variants of interest (VOIs) requires robust surveillance systems, including integrated genome sequencing with a well-defined sampling and sequencing strategy to ensure representativeness and reliability of findings” (ECDC, 2021, 2022b).
The EDCD released guidelines on the use of antibody tests for SARS-CoV-2 in 2022. The key messages are:
- “At present, antibody tests are mostly used in research studies (mainly sero-epidemiological) at population level rather than for individual diagnosis of COVID-19 cases.
- A positive antibody test result can indicate a previous infection or vaccination but cannot be used to determine whether an individual is currently infectious or protected against infection.
- In the absence of a positive diagnostic test result, antibody tests cannot determine the time of infection.
- The antibody titres that correlate with protection from infection are currently unknown.
- There are a variety of antibody tests available and it is extremely difficult to compare their results due to the diversity and lack of standardisation.
- Antibody tests that target the spike protein are unable to distinguish between those who have been previously infected and those who have received at least one dose of a SARS-CoV-2 vaccine.
- There is a risk that the antibodies detected by the commercial tests currently in use will not prevent infection with newly emerging SARS-CoV-2 variants” (ECDC, 2022a).
American Academy of Pediatrics (AAP)
The AAP lists the most common scenarios for testing as symptomatic patients; patients who are asymptomatic but had exposure to a person with confirmed or probable COVID-19 infection; and patients who required screening as part of local public health, school, or workplace requirement. The AAP notes that a person’s vaccination status may be a factor in decision-making concerning the need for screening (AAP, 2022).
Additionally, the AAP says that for patients who have symptoms, both NAATs (such as PCR testing) and antigen tests can be used. A positive result indicates a SARS-CoV-2 infection on either PCR or antigen diagnostics. That said, for a patient with a negative antigen result, a provider may repeat the antigen test at 48 hours per FDA guidance (AAP, 2022).
For purposes of testing symptomatic children who have recently had confirmed infections within three months, the AAP says providers should consider the possibility of a false-positive result. Especially using PCR tests and other NAAT tests, as these may remain positive from deposited viral genetic material for several months after an active infection. The AAP notes, “In a child with known exposure and compatible symptoms, there may be situations in which it is reasonable to retest within the 90-day window. If testing is performed within that window, antigen testing is generally preferable to NAATs because of the potential for positive NAAT results attributable to prior infection” (AAP, 2022).
Further, the AAP previously stated in 2020-2021 guidance that antibody (serologic) tests “can provide evidence of previous infection with SARS-CoV-2 but are not useful for the diagnosis of acute infection. A positive antibody test result does not prove that a patient has protection against SARS-CoV-2, although the FDA and vaccine companies use serologic testing as a marker for immunogenicity and protection from SARS-CoV-2 infection. Thus, these tests should not be used to make decisions on grouping people in classrooms or other facilities at this time, and individuals with positive antibody tests should continue to adhere to guidelines about masking, social distancing, and other preventive measures” (AAP, 2022).
The AAP has also included some comments and discussion on Multisystem Inflammatory Syndrome in Children (MIS-C). MIS-C has been observed to have some association with COVID-19, and patients with this syndrome have been observed to test positive “far more often” for past SARS-CoV-2 infection (i.e., antibody testing) than acute infection (RT-PCR or antigen test). The Council of State and Territorial Epidemiologists (CSTE) and CDC defines an MIS-C case by the following criteria:
“An individual aged < 21 years and in the absence of a more likely alternative diagnosis:
- Subjective or documented fever (T > 38.0° C)
- Clinical severity requiring hospitalization or resulting in death
- C-reactive protein (CRP) >3.0 mg/dL
- New onset manifestations of >2 of the following categories:
- Cardiac: coronary artery dilatation/aneurysm, left ventricular ejection fraction < 55%, or troponin elevated above normal
- Shock
- Mucocutaneous: rash, oral mucosal inflammation, conjunctivitis/conjunctival injection or extremity findings (erythema, edema)
- Gastrointestinal: abdominal pain, vomiting or diarrhea
- Hematologic: platelet count < 150,000/µL, absolute lymphocyte count < 1000/µL
- Detection of SARS-CoV-2 nucleic acid/antigen up to 60 days prior to or during hospitalization or in a postmortem specimen, OR detection of antibody associated with current illness, OR close contact with a confirmed/probable COVID-19 case in the 60 days prior to hospitalization” (AAP, 2023).
The CDC delineates a testing algorithm for MIS-C in the outpatient or emergency department setting as follows:
· “Evaluate a child with persistent fever (≥ 3 days) who is moderately to severely ill with clinical signs of organ dysfunction (eg, gastrointestinal, respiratory, cardiac, mucocutaneous or hematologic). Initial evaluation should include measurement of vital signs, assessment of perfusion and oxygen saturation. Early consultation and coordination with the nearest pediatric infectious disease and rheumatology specialist and pediatric referral center for optimal testing and management should be considered. Laboratory screening for systemic inflammation may be considered and initial lab screenings may include complete blood cell count (CBC) with differential, urine analysis, ESR, and CRP, with the addition of ferritin, LDH, comprehensive metabolic panel, pro-BNP, troponin and fibrinogen depending on initial clinical suspicion and/or evidence of inflammation on initial lab screening. Note that none of these laboratory studies is specific for the diagnosis of MIS-C, so even if there is evidence of significant systemic inflammation, alternative diagnoses must still be considered (eg, pyelonephritis, appendicitis)” (AAP, 2023).
For the evaluation of severely ill appearing or hemodynamically fragile patients, they propose that:
“Severely ill-appearing patients and those in compensated shock or shock should be evaluated and treated in the emergency department/critical care setting. Transfer to a referral center should be arranged. Laboratory tests, as described above, should be performed for initial evaluation regardless of duration of fever. Consultation with pediatric subspecialists (infectious diseases, cardiology, rheumatology) at a local or regional pediatric referral center should be initiated but should not delay transfer to a referral center” (AAP, 2023).
Testing for hospitalized children is delineated below.
“Any child sick enough to warrant admission for fever, abdominal pain, diarrhea and/or organ dysfunction in whom MIS-C is suspected should be cared for in a hospital with tertiary pediatric/cardiac intensive care units. Although decisions about additional testing will be made by the multidisciplinary team managing the patient, pediatricians can prepare families for an expanded laboratory and cardiac workup that may include:
- Chest radiograph, EKG and troponin. If any of these or physical examination is abnormal, then consult with pediatric cardiology and consider additional diagnostic testing for myocardial injury (echocardiogram and/or cardiac MRI).
- Expanded laboratory tests including pro-BNP, triglycerides, creatine kinase, amylase, blood and urine culture, D-dimer, prothrombin time/partial thromboplastin time (PT/PTT), INR, CRP, ferritin, LDH, comprehensive metabolic panel and fibrinogen, if not already conducted.
- In all cases, COVID-19 testing should be performed with RT-PCR assay and serologic testing. Later serology may be needed if all are negative initially. Serologic tests must be sent prior to administration of intravenous immunoglobulin (IVIG)” (AAP, 2023).
American College of Rheumatology (ACR)
The ACR published guidance regarding MIS-C associated with COVID-19. In it, they list SARS-CoV-2 IgG, IgM, and IgA as part of the diagnostic pathway for MIS-C (Henderson, Canna, Friedman, Gorelik, Lapidus, Bassiri, Behrens, Ferris, Kernan, Schulert, Seo, MB, et al., 2020).
In a December 5, 2020 update of the above guidelines, the ACR states that ESR, CRP, and testing for SARS-CoV-2 (by PCR or serology) should be considered a “tier 1” (first-line evaluation) for MIS-C (Henderson, Canna, Friedman, Gorelik, Lapidus, Bassiri, Behrens, Ferris, Kernan, Schulert, Seo, Son, et al., 2020).
In a February 3, 2022 update of the above guideline, the ACR added new information concerning immunomodulatory treatment in MIS-C, hyperinflammation in COVID-19, as well as statements on thrombotic risk and anticoagulation in MIS-C (Henderson et al., 2022).
References
- AAP. (2022, February 28). COVID-19 Testing Guidance. https://services.aap.org/en/pages/2019-novel-coronavirus-covid-19-infections/clinical-guidance/covid-19-testing-guidance/
- AAP. (2023, February 8). Multisystem Inflammatory Syndrome in Children (MIS-C) Interim Guidance https://www.aap.org/en/pages/2019-novel-coronavirus-covid-19-infections/clinical-guidance/multisystem-inflammatory-syndrome-in-children-mis-c-interim-guidance/
- AMA. (2020, May 14, 2020). Serological testing for SARS-CoV-2 antibodies. American Medical Association. https://www.ama-assn.org/delivering-care/public-health/serological-testing-sars-cov-2-antibodies
- ASA, & APSF. (2022, December 21, 2022). ASA and APSF Updated Statement on Perioperative Testing for SARS-CoV-2 in the Asymptomatic Patient. https://www.apsf.org/news-updates/asa-and-apsf-updated-statement-on-perioperative-testing-for-sars-cov-2-in-the-asymptomatic-patient/
- Backer, J. A., Klinkenberg, D., & Wallinga, J. (2020). Incubation period of 2019 novel coronavirus (2019-nCoV) infections among travellers from Wuhan, China, 20-28 January 2020. Euro Surveill, 25(5). https://doi.org/10.2807/1560-7917.Es.2020.25.5.2000062
- Baum, S. G. (2020). Adult Multisystem Inflammatory Syndrome Associated with COVID-19. NEJM. https://www.jwatch.org/na52622/2020/10/21/adult-multisystem-inflammatory-syndrome-associated-with BD Veritor. (2020). Veritor™ System https://www.fda.gov/media/139755/download
- Bezerra, M. F., Machado, L. C., De Carvalho, V., Docena, C., Brandão-Filho, S. P., Ayres, C. F. J., Paiva, M. H. S., & Wallau, G. L. (2021). A Sanger-based approach for scaling up screening of SARS-CoV-2 variants of interest and concern. Infect Genet Evol, 92, 104910. https://doi.org/10.1016/j.meegid.
2021.104910 - BioFire. (2020). BioFire® Respiratory Panel 2.1 (RP2.1). FDA. https://www.fda.gov/media/137583/download BioGerm. (2024). 2019-nCoV nucleic acid detection kit. https://www.bio-germ.com/
- BioSpace. (2020, 8/20/20). Quidel to Update Packaging of Point-of-Care Sofia® SARS Antigen Test for COVID-19 to Include Either Nasal or Nasopharyngeal Swabs.
- BodiTechMed. (2024). AFIAS COVID-19 Ab. http://www.boditech.co.kr/eng/board/news/board_view.asp?num = 30109
- Caturegli, G., Materi, J., Howard, B. M., & Caturegli, P. (2020). Clinical Validity of Serum Antibodies to SARS-CoV-2 : A Case-Control Study. Ann Intern Med, 173(8), 614-622. https://doi.org/10.7326/m20-2889
- CDC. (2020, Febuary 15). Human Coronavirus Types. CDC. Retrieved 05/15/2020 from https://archive.cdc.gov/#/details?url = https://www.cdc.gov/coronavirus/types.html CDC. (2024a, June 13, 2024). About COVID-19. https://www.cdc.gov/covid/about/index.html
- CDC. (2024b, January 8, 2024). About Whole Genome Sequencing. https://www.cdc.gov/pulsenet/php/wgs/
- CDC. (2024c, October 31, 2024). Interim Clinical Considerations for Use of COVID-19 Vaccines Currently Approved or Authorized in the United States. https://www.cdc.gov/vaccines/covid-19/clinical-considerations/interim-considerations-us.html CDC. (2024d, July 11, 2024). Long COVID Basics. https://www.cdc.gov/covid/long-term-effects/
- CDC. (2024e, May 29, 2024). Multisystem Inflammatory Syndrome: Case Definitions and Reporting. https://www.cdc.gov/mis/hcp/case-definition-reporting/index.html
- CDC. (2024f, August 29, 2024). Overview of Testing for SARS-CoV-2. https://www.cdc.gov/covid/hcp/clinical-care/overview-testing-sars-cov-2.html CDC. (2024g, June 25, 2024). Symptoms of COVID-19. https://www.cdc.gov/covid/signs-symptoms/
- CDC. (2024h, August 24, 2024). Testing for COVID-19. https://www.cdc.gov/covid/testing/index.html
- Cevik, M., Tate, M., Lloyd, O., Maraolo, A. E., Schafers, J., & Ho, A. (2021). SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: a systematic review and meta-analysis. The Lancet Microbe, 2(1), E13-E22. https://doi.org/10.1016/S2666-5247(20)30172-5
- Chan, J. F., Yip, C. C., To, K. K., Tang, T. H., Wong, S. C., Leung, K. H., Fung, A. Y., Ng, A. C., Zou, Z., Tsoi, H. W., Choi, G. K., Tam, A. R., Cheng, V. C., Chan, K. H., Tsang, O. T., & Yuen, K. Y. (2020). Improved Molecular Diagnosis of COVID-19 by the Novel, Highly Sensitive and Specific COVID-19-RdRp/Hel Real-Time Reverse Transcription-PCR Assay Validated In Vitro and with Clinical Specimens. J Clin Microbiol, 58(5). https://doi.org/10.1128/jcm.00310-20
- Chau, N. V. V., Hong, N. T. T., Ngoc, N. M., Anh, N. T., Trieu, H. T., Nhu, L. N. T., Yen, L. M., Minh, N. N. Q., Phong, N. T., Truong, N. T., Huong, L. T. T., Tu, T. N. H., Hung, L. M., Thanh, T. T., Dung, N. T., Dung, N. T., Thwaites, G., Van Tan, L., & for, O. C.-r. g. (2021). Rapid whole-genome sequencing to inform COVID-19 outbreak response in Vietnam. The Journal of infection, 82(6), 276-316. https://doi.org/10.1016/j.jinf.2021.03.017
- Churiwal, M., Lin, K. D., Khan, S., Chhetri, S., Muller, M. S., Tompkins, K., Smith, J., Litel, C., Whittelsey, M., Basham, C., Rapp, T., Cerami, C., Premkumar, L., & Lin, J. T. (2021). Assessment of the Field Utility of a Rapid Point-of-Care Test for SARS-CoV-2 Antibodies in a Household Cohort. Am J Trop Med Hyg, 106(1), 156-159. https://doi.org/10.4269/ajtmh.21-0592
- Corman, V. M., Lienau, J., & Witzenrath, M. (2019). [Coronaviruses as the cause of respiratory infections]. Internist (Berl), 60(11), 1136-1145. https://doi.org/10.1007/s00108-019-00671-5 (Coronaviren als Ursache respiratorischer Infektionen.)
- Cucinotta, D., & Vanelli, M. (2020). WHO Declares COVID-19 a Pandemic. Acta Biomed, 91(1), 157-160. https://doi.org/10.23750/abm.v91i1.9397
- Dao Thi, V. L., Herbst, K., Boerner, K., Meurer, M., Kremer, L. P. M., Kirrmaier, D., Freistaedter, A., Papagiannidis, D., Galmozzi, C., Stanifer, M. L., Boulant, S., Klein, S., Chlanda, P., Khalid, D., Barreto Miranda, I., Schnitzler, P., Kräusslich, H.-G., Knop, M., & Anders, S. (2020). A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Science Translational Medicine, 12(556), eabc7075. https://doi.org/10.1126/scitranslmed.abc7075
- DeBiasi, R. L., Song, X., Delaney, M., Bell, M., Smith, K., Pershad, J., Ansusinha, E., Hahn, A., Hamdy, R., Harik, N., Hanisch, B., Jantausch, B., Koay, A., Steinhorn, R., Newman, K., & Wessel, D. (2020). Severe COVID-19 in Children and Young Adults in the Washington, DC Metropolitan Region. J Pediatr. https://doi.org/10.1016/j.jpeds.2020.05.007
- Diao, B., Wen, K., Chen, J., Liu, Y., Yuan, Z., Han, C., Chen, J., Pan, Y., Chen, L., Dan, Y., Wang, J., Chen, Y., Deng, G., Zhou, H., & Wu, Y. (2020). Diagnosis of Acute Respiratory Syndrome Coronavirus 2 Infection by Detection of Nucleocapsid Protein. medRxiv, 2020.2003.2007.20032524. https://doi.org/10.1101/2020.03.07.20032524
- Dighe, K., Moitra, P., Alafeef, M., Gunaseelan, N., & Pan, D. (2022). A rapid RNA extraction-free lateral flow assay for molecular point-of-care detection of SARS-CoV-2 augmented by chemical probes. Biosensors and Bioelectronics, 200, 113900. https://doi.org/10.1016/j.bios.
2021.113900 - ECDC. (2021, May 3). Guidance for representative and targeted genomic SARS-CoV-2 monitoring. https://www.ecdc.europa.eu/en/publications-data/guidance-representative-and-targeted-genomic-sars-cov-2-monitoring
- ECDC. (2022a). Considerations for the use of antibody tests for SARS-CoV-2 – first update. https://www.ecdc.europa.eu/en/publications-data/use-antibody-tests-sars-cov-2
- ECDC. (2022b, December 15). Testing strategies for SARS-CoV-2. https://www.ecdc.europa.eu/en/covid-19/surveillance/testing-strategies
- ECDC. (2023, 03/22/2022). Diagnostic testing and screening for SARS-CoV-2. European Centre for Disease Prevention and Control. Retrieved 04/18/2022 from https://www.ecdc.europa.eu/en/covid-19/latest-evidence/diagnostic-testing
- EpitopeDiagnostics. (2024). EDI™ Novel Coronavirus COVID-19 ELISA Kits. http://www.epitopediagnostics.com/covid-19-elisa
- Espejo, A. P., Akgun, Y., Al Mana, A. F., Tjendra, Y., Millan, N. C., Gomez-Fernandez, C., & Cray, C. (2020). Review of Current Advances in Serologic Testing for COVID-19. Am J Clin Pathol, 154(3), 293-304. https://doi.org/10.1093/ajcp/aqaa112
- FDA. (2020). Coronavirus (COVID-19) Update: FDA Issues First Emergency Authorization for Sample Pooling in Diagnostic Testing. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-first-emergency-authorization-sample-pooling-diagnostic FDA. (2021a, May 11). Coronavirus (COVID-19) Update: 10/15/21. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-101521
- FDA. (2021b, April 22). Illumina COVIDSeq Test. https://www.fda.gov/media/138778/download
- FDA. (2021c, August 5). Influenza SARS-CoV-2 (Flu SC2) Multiplex Assay. https://www.fda.gov/media/139744/download FDA. (2023a, November 8). In Vitro Diagnostics EUAs. https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/vitro-diagnostics-euas
- FDA. (2023b). Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised). FDA. Retrieved 04/20/2022 from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised
- FDA. (2023c). Transition Plan for Medical Devices Issued Emergency Use Authorizations (EUAs) Related to Coronavirus Disease 2019 (COVID-19). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/transition-plan-medical-devices-issued-emergency-use-authorizations-euas-related-coronavirus-disease
- FDA. (2023d). Transition Plan for Medical Devices That Fall Within Enforcement Policies Issued During the Coronavirus Disease 2019 (COVID-19) Public Health Emergency. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/transition-plan-medical-devices-fall-within-enforcement-policies-issued-during-coronavirus-disease
- FDA. (2024a, July 1). ACCELERATED EMERGENCY USE AUTHORIZATION (EUA) SUMMARY SARS-CoV-2 RT-PCR Assay. https://www.fda.gov/media/141192/download
- FDA. (2024b, August 6). CDC Influenza SARS-CoV-2 (Flu SC2) Multiplex Assay. https://www.fda.gov/media/139743/download
- FDA. (2024c, August 27). Emergency Use Authorization. Retrieved 04/20/2022 from https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization
- Fox, T., Geppert, J., Dinnes, J., Scandrett, K., Bigio, J., Sulis, G., Hettiarachchi, D., Mathangasinghe, Y., Weeratunga, P., Wickramasinghe, D., Bergman, H., Buckley, B. S., Probyn, K., Sguassero, Y., Davenport, C., Cunningham, J., Dittrich, S., Emperador, D., Hooft, L., . . . Deeks, J. J. (2022). Antibody tests for identification of current and past infection with SARS-CoV-2. Cochrane Database Syst Rev, 11(11), Cd013652. https://doi.org/10.1002/14651858.CD013652.pub2
- Gandhi, R. (2024, March 19). COVID-19: Clinical features. Wolter Kluwer. Retrieved 08/19/2020 from https://www.uptodate.com/contents/covid-19-clinical-features
- GenMark Diagnostics. (2024). ePlex Respiratory Pathogen Panel 2. https://www.fda.gov/media/142902/download
- Greninger, A. L., Dien Bard, J., Colgrove, R. C., Graf, E. H., Hanson, K. E., Hayden, M. K., Humphries, R. M., Lowe, C. F., Miller, M. B., Pillai, D. R., Rhoads, D. D., Yao, J. D., & Lee, F. M. (2022). Clinical and Infection Prevention Applications of Severe Acute Respiratory Syndrome Coronavirus 2 Genotyping: An Infectious Diseases Society of America/American Society for Microbiology Consensus Review Document. Clin Infect Dis, 74(8), 1496-1502. https://doi.org/10.1093/cid/ciab761
- Griffin, D. (2020, December 31). Viral Load as a Predictor of COVID-19 Patient Outcomes. https://www.cuimc.columbia.edu/news/viral-load-predictor-covid-19-patient-outcomes
- Guo, L., Ren, L., Yang, S., Xiao, M., Chang, D., Yang, F., Dela Cruz, C. S., Wang, Y., Wu, C., Xiao, Y., Zhang, L., Han, L., Dang, S., Xu, Y., Yang, Q.-W., Xu, S.-Y., Zhu, H.-D., Xu, Y.-C., Jin, Q., . . . Wang, J. (2020). Profiling Early Humoral Response to Diagnose Novel Coronavirus Disease (COVID-19). Clinical Infectious Diseases. https://doi.org/10.1093/cid/ciaa310
- Helix. (2020, 8/6/20). Helix COVID-19 NGS Test. Retrieved 8/20/20 from https://www.fda.gov/media/140917/download
- Henderson, L. A., Canna, S. W., Friedman, K. G., Gorelik, M., Lapidus, S. K., Bassiri, H., Behrens, E. M., Ferris, A., Kernan, K. F., Schulert, G. S., Seo, P., MB, F. S., Tremoulet, A. H., Yeung, R. S. M., Mudano, A. S., Turner, A. S., Karp, D. R., & Mehta, J. J. (2020). American College of Rheumatology Clinical Guidance for Multisystem Inflammatory Syndrome in Children Associated With SARS-CoV-2 and Hyperinflammation in Pediatric COVID-19: Version 1. Arthritis Rheumatol. https://doi.org/10.1002/art.41454
- Henderson, L. A., Canna, S. W., Friedman, K. G., Gorelik, M., Lapidus, S. K., Bassiri, H., Behrens, E. M., Ferris, A., Kernan, K. F., Schulert, G. S., Seo, P., Son, M. B. F., Tremoulet, A. H., Yeung, R. S. M., Mudano, A. S., Turner, A. S., Karp, D. R., & Mehta, J. J. (2020). American College of Rheumatology Clinical Guidance for Pediatric Patients with Multisystem Inflammatory Syndrome in Children (MIS-C) Associated with SARS-CoV-2 and Hyperinflammation in COVID-19. Version 2. Arthritis Rheumatol. https://doi.org/10.1002/art.41616
- Henderson, L. A., Canna, S. W., Friedman, K. G., Gorelik, M., Lapidus, S. K., Bassiri, H., Behrens, E. M., Kernan, K. F., Schulert, G. S., Seo, P., Son, M. B. F., Tremoulet, A. H., VanderPluym, C., Yeung, R. S. M., Mudano, A. S., Turner, A. S., Karp, D. R., & Mehta, J. J. (2022). American College of Rheumatology Clinical Guidance for Multisystem Inflammatory Syndrome in Children Associated With SARS–CoV-2 and Hyperinflammation in Pediatric COVID-19: Version 3. Arthritis & Rheumatology, 74(4), e1-e20. https://doi.org/10.1002/art.42062
- Hirotsu, Y., Maejima, M., Shibusawa, M., Nagakubo, Y., Hosaka, K., Amemiya, K., Sueki, H., Hayakawa, M., Mochizuki, H., Tsutsui, T., Kakizaki, Y., Miyashita, Y., Yagi, S., Kojima, S., & Omata, M. (2020). Comparison of Automated SARS-CoV-2 Antigen Test for COVID-19 Infection with Quantitative RT-PCR using 313 Nasopharyngeal Swabs Including from 7 Serially Followed Patients. Int J Infect Dis. https://doi.org/10.1016/j.ijid.2020.08.029
- Hogan, C. A., Sahoo, M. K., & Pinsky, B. A. (2020). Sample Pooling as a Strategy to Detect Community Transmission of SARS-CoV-2. Jama, 323(19), 1967-1969. https://doi.org/10.1001/jama.2020.5445
- Hulick, P. (2024, October 25 2024). Next-generation DNA sequencing (NGS): Principles and clinical applications. Wolters Kluwer. https://www.uptodate.com/contents/next-generation-dna-sequencing-ngs-principles-and-clinical-applications
- IDSA. (2023). Infectious Diseases Society of America Guidelines on the Diagnosis of COVID-19: Molecular Diagnostic Testing. IDSA. Retrieved 05/13/2020 from https://www.idsociety.org/practice-guideline/covid-19-guideline-diagnostics/
- IDSA. (2024, Febuary 9). Infectious Diseases Society of America Guidelines on the Diagnosis of COVID-19: Serologic Testing. https://www.idsociety.org/practice-guideline/covid-19-guideline-serology/
- Jones, V. G., Mills, M., Suarez, D., Hogan, C. A., Yeh, D., Bradley Segal, J., Nguyen, E. L., Barsh, G. R., Maskatia, S., & Mathew, R. (2020). COVID-19 and Kawasaki Disease: Novel Virus and Novel Case. Hosp Pediatr. https://doi.org/10.1542/hpeds.2020-0123
- Kawasuji, H., Takegoshi, Y., Kaneda, M., Ueno, A., Miyajima, Y., Kawago, K., Fukui, Y., Yoshida, Y., Kimura, M., Yamada, H., Sakamaki, I., Tani, H., Morinaga, Y., & Yamamoto, Y. (2020). Transmissibility of COVID-19 depends on the viral load around onset in adult and symptomatic patients. PLOS ONE, 15(12), e0243597. https://doi.org/10.1371/journal.pone.0243597
- Ko, J. H., Joo, E. J., Park, S. J., Baek, J. Y., Kim, W. D., Jee, J., Kim, C. J., Jeong, C., Kim, Y. J., Shon, H. J., Kang, E. S., Choi, Y. K., & Peck, K. R. (2020). Neutralizing Antibody Production in Asymptomatic and Mild COVID-19 Patients, in Comparison with Pneumonic COVID-19 Patients. J Clin Med, 9(7). https://doi.org/10.3390/jcm9072268
- Kontou, P. I., Braliou, G. G., Dimou, N. L., Nikolopoulos, G., & Bagos, P. G. (2020). Antibody Tests in Detecting SARS-CoV-2 Infection: A Meta-Analysis. Diagnostics (Basel), 10(5). https://doi.org/10.3390/diagnostics10050319
- Kweon, O. J., Lim, Y. K., Kim, H. R., Kim, M. C., Choi, S. H., Chung, J. W., & Lee, M. K. (2020). Antibody kinetics and serologic profiles of SARS-CoV-2 infection using two serologic assays. PLOS ONE, 15(10), e0240395. https://doi.org/10.1371/journal.pone.0240395
- LabCorp. (2022a, June 21). ACCELERATED EMERGENCY USE AUTHORIZATION (EUA) SUMMARY. Retrieved 8/21/20 from https://www.fda.gov/media/136151/download
- LabCorp. (2022b, June 21). ACCELERATED EMERGENCY USE AUTHORIZATION (EUA) SUMMARY COVID-19 RT-PCR TEST (LABORATORY CORPORATION OF AMERICA). FDA. Retrieved 04/26/2020 from https://www.fda.gov/media/136151/download
- Lambert-Niclot, S., Cuffel, A., Le Pape, S., Vauloup-Fellous, C., Morand-Joubert, L., Roque-Afonso, A. M., Le Goff, J., & Delaugerre, C. (2020). Evaluation of a Rapid Diagnostic Assay for Detection of SARS-CoV-2 Antigen in Nasopharyngeal Swabs. J Clin Microbiol, 58(8). https://doi.org/10.1128/jcm.00977-20
- Li, M., Wei, R., Yang, Y., He, T., Shen, Y., Qi, T., Han, T., Song, Z., Zhu, Z., Ma, X., Lin, Y., Yuan, Y., Zhao, K., Lu, H., & Zhou, X. (2021). Comparing SARS-CoV-2 Testing in Anterior Nasal Vestibular Swabs vs. Oropharyngeal Swabs. Front Cell Infect Microbiol, 11, 653794. https://doi.org/10.3389/fcimb.
2021.653794 - Li, Y., Yao, L., Li, J., Chen, L., Song, Y., Cai, Z., & Yang, C. (2020). Stability issues of RT-PCR testing of SARS-CoV-2 for hospitalized patients clinically diagnosed with COVID-19. Journal of medical virology, 92(7), 903-908. https://doi.org/10.1002/jmv.25786
- Lippi, G., Simundic, A. M., & Plebani, M. (2020). Potential preanalytical and analytical vulnerabilities in the laboratory diagnosis of coronavirus disease 2019 (COVID-19). Clin Chem Lab Med. https://doi.org/10.1515/cclm-2020-0285
- Lisboa Bastos, M., Tavaziva, G., Abidi, S. K., Campbell, J. R., Haraoui, L. P., Johnston, J. C., Lan, Z., Law, S., MacLean, E., Trajman, A., Menzies, D., Benedetti, A., & Ahmad Khan, F. (2020). Diagnostic accuracy of serological tests for covid-19: systematic review and meta-analysis. Bmj, 370, m2516. https://doi.org/10.1136/bmj.m2516
- Loeffelholz, M. J., & Tang, Y.-W. (2020). Laboratory diagnosis of emerging human coronavirus infections – the state of the art. Emerging Microbes & Infections, 9(1), 747-756. https://doi.org/10.1080/22221751.2020.1745095
- Lu, Y., Li, L., Ren, S., Liu, X., Zhang, L., Li, W., & Yu, H. (2020). Comparison of the diagnostic efficacy between two PCR test kits for SARS-CoV-2 nucleic acid detection. Journal of Clinical Laboratory Analysis, 34(10), e23554. https://doi.org/10.1002/jcla.23554
- Ludwig, S., & Zarbock, A. (2020). Coronaviruses and SARS-CoV-2: A Brief Overview. Anesth Analg. https://doi.org/10.1213/ane.0000000000004845 LumiraDx. (2020). SARS-CoV-2 Ag Test. https://www.fda.gov/media/141304/download
- Mak, G. C., Cheng, P. K., Lau, S. S., Wong, K. K., Lau, C. S., Lam, E. T., Chan, R. C., & Tsang, D. N. (2020). Evaluation of rapid antigen test for detection of SARS-CoV-2 virus. J Clin Virol, 129, 104500. https://doi.org/10.1016/j.jcv.
2020.104500 - Mawhorter, M. E., Nguyen, P., Goldsmith, M., Owens, R. G., Baer, B., & Raman, J. D. (2022). Diagnostic yield and costs associated with a routine pre-operative COVID-19 testing algorithm for asymptomatic patients prior to elective surgery. Am J Clin Exp Urol, 10(5), 341-344.
- Mboumba Bouassa, R.-S., Tonen-Wolyec, S., Veyer, D., Péré, H., & Bélec, L. (2022). Analytical performances of the AMPLIQUICK® Respiratory Triplex assay for simultaneous detection and differentiation of SARS-CoV-2, influenza A/B and respiratory syncytial viruses in respiratory specimens. PLOS ONE, 17(1), e0262258. https://doi.org/10.1371/journal.pone.0262258
- Morell, A., Skvaril, F., Noseda, G., & Barandun, S. (1973). Metabolic properties of human IgA subclasses. Clin Exp Immunol, 13(4), 521-528. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1553728/
- Morris, S. B., Schwartz, N. G., Patel, P., Abbo, L., Beauchamps, L., Balan, S., Lee, E. H., Paneth-Pollak, R., Geevarughese, A., Lash, M. K., Dorsinville, M. S., Ballen, V., Eiras, D. P., Newton-Cheh, C., Smith, E., Robinson, S., Stogsdill, P., Lim, S., Fox, S. E., . . . Godfred-Cato, S. (2020). Case Series of Multisystem Inflammatory Syndrome in Adults Associated with SARS-CoV-2 Infection - United Kingdom and United States, March-August 2020. MMWR Morb Mortal Wkly Rep, 69(40), 1450-1456. https://doi.org/10.15585/mmwr.mm6940e1
- Nagura-Ikeda, M., Imai, K., Tabata, S., Miyoshi, K., Murahara, N., Mizuno, T., Horiuchi, M., Kato, K., Imoto, Y., Iwata, M., Mimura, S., Ito, T., Tamura, K., & Kato, Y. (2020). Clinical evaluation of self-collected saliva by RT-qPCR, direct RT-qPCR, RT-LAMP, and a rapid antigen test to diagnose COVID-19. J Clin Microbiol. https://doi.org/10.1128/jcm.01438-20
- NIH. (2024a, May 20). Clinical Spectrum of SARS-CoV-2 Infection. National Institutes of Health. https://medlineplus.gov/covid19coronavirusdisease2019.html
- NIH. (2024b, April 1). Testing for SARS-CoV-2 Infection. National Institutes of Health. https://medlineplus.gov/covid19testing.html
- Okba, N. M. A., Müller, M. A., Li, W., Wang, C., GeurtsvanKessel, C. H., Corman, V. M., Lamers, M. M., Sikkema, R. S., de Bruin, E., Chandler, F. D., Yazdanpanah, Y., Le Hingrat, Q., Descamps, D., Houhou-Fidouh, N., Reusken, C., Bosch, B. J., Drosten, C., Koopmans, M. P. G., & Haagmans, B. L. (2020). Severe Acute Respiratory Syndrome Coronavirus 2-Specific Antibody Responses in Coronavirus Disease 2019 Patients. Emerg Infect Dis, 26(7). https://doi.org/10.3201/eid
2607.200841 - Oude Munnink, B. B., Nieuwenhuijse, D. F., Stein, M., O’Toole, Á., Haverkate, M., Mollers, M., Kamga, S. K., Schapendonk, C., Pronk, M., Lexmond, P., van der Linden, A., Bestebroer, T., Chestakova, I., Overmars, R. J., van Nieuwkoop, S., Molenkamp, R., van der Eijk, A. A., GeurtsvanKessel, C., Vennema, H., . . . The Dutch-Covid-19 response, t. (2020). Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nature Medicine, 26(9), 1405-1410. https://doi.org/10.1038/s41591-020-0997-y
- Padoan, A., Cosma, C., Sciacovelli, L., Faggian, D., & Plebani, M. (2020). Analytical performances of a chemiluminescence immunoassay for SARS-CoV-2 IgM/IgG and antibody kinetics. Clin Chem Lab Med. https://doi.org/10.1515/cclm-2020-0443
- Peacock, W. F., Soto-Ruiz, K. M., House, S. L., Cannon, C. M., Headden, G., Tiffany, B., Motov, S., Merchant-Borna, K., Chang, A. M., Pearson, C., Patterson, B. W., Jones, A. E., Miller, J., Varon, J., Bastani, A., Clark, C., Rafique, Z., Kea, B., Eppensteiner, J., . . . Young, S. (2022). Utility of COVID-19 antigen testing in the emergency department. Journal of the American College of Emergency Physicians Open, 3(1), e12605. https://doi.org/https://doi.org/10.1002/emp2.12605
- Pfefferle, S., Reucher, S., Nörz, D., & Lütgehetmann, M. (2020). Evaluation of a quantitative RT-PCR assay for the detection of the emerging coronavirus SARS-CoV-2 using a high throughput system. Euro Surveill, 25(9). https://doi.org/10.2807/1560-7917.Es.2020.25.9.2000152
- Poljak, M., Korva, M., Gašper, N. K., Komloš, K. F., Sagadin, M., Uršič, T., Županc, T. A., Petrovec, M., & McAdam, A. J. (2020). Clinical Evaluation of the cobas SARS-CoV-2 Test and a Diagnostic Platform Switch during 48 Hours in the Midst of the COVID-19 Pandemic. Journal of Clinical Microbiology, 58(6), e00599-00520. https://doi.org/doi:10.1128/JCM.00599-20
- Poplar. (2020). EMERGENCY USE AUTHORIZATION (EUA) SUMMARY OF THE POPLAR SARS-COV-2 TMA POOLING ASSAY. https://www.fda.gov/media/140792/download
- Qiagen GmbH. (2021, July). QIAstat-Dx® Respiratory SARS-CoV2 Panel Instructions for Use (Handbook). FDA. Retrieved 04/27/2020 from https://www.fda.gov/media/136571/download
- Quidel Corporation. (2020, 05/2020). Sofia 2 SARS Antigen FIA. FDA. Retrieved 05/12/2020 from https://www.fda.gov/media/137885/download Ryding, S. (2020, June 24). What is Viral Load? Retrieved January 31 from https://www.news-medical.net/health/What-is-Viral-Load.aspx
- SansureBiotech. (2022, March 25). Novel Coronavirus (2019-nCoV) Nucleic Acid Diagnostic Kit (PCR-Fluorescence Probing). https://www.fda.gov/media/137651/download
- Scohy, A., Anantharajah, A., Bodéus, M., Kabamba-Mukadi, B., Verroken, A., & Rodriguez-Villalobos, H. (2020). Low performance of rapid antigen detection test as frontline testing for COVID-19 diagnosis. J Clin Virol, 129, 104455. https://doi.org/10.1016/j.jcv.
2020.104455 - Seo, G., Lee, G., Kim, M. J., Baek, S. H., Choi, M., Ku, K. B., Lee, C. S., Jun, S., Park, D., Kim, H. G., Kim, S. J., Lee, J. O., Kim, B. T., Park, E. C., & Kim, S. I. (2020). Rapid Detection of COVID-19 Causative Virus (SARS-CoV-2) in Human Nasopharyngeal Swab Specimens Using Field-Effect Transistor-Based Biosensor. ACS Nano, 14(4), 5135-5142. https://doi.org/10.1021/acsnano.0c02823
- Sri Santosh, T., Parmar, R., Anand, H., Srikanth, K., & Saritha, M. (2020). A Review of Salivary Diagnostics and Its Potential Implication in Detection of Covid-19. Cureus, 12(4), e7708. https://doi.org/10.7759/cureus.7708
- Talbot, T. R., Hayden, M. K., Yokoe, D. S., Malani, A. N., Amer, H. A., Kalu, I. C., Logan, L. K., Moehring, R. W., Munoz-Price, S., Palmore, T. N., Weber, D. J., Wright, S. B., & Trustees, S. B. o. (2023). Asymptomatic screening for severe acute respiratory coronavirus virus 2 (SARS-CoV-2) as an infection prevention measure in healthcare facilities: Challenges and considerations. Infect Control Hosp Epidemiol, 44(1), 2-7. https://doi.org/10.1017/ice.2022.295
- Taylor, J., Carter, R. J., Lehnertz, N., Kazazian, L., Sullivan, M., Wang, X., Garfin, J., Diekman, S., Plumb, M., Bennet, M. E., Hale, T., Vallabhaneni, S., Namugenyi, S., Carpenter, D., Turner-Harper, D., Booth, M., Coursey, E. J., Martin, K., McMahon, M., . . . Lynfield, R. (2020). Serial Testing for SARS-CoV-2 and Virus Whole Genome Sequencing Inform Infection Risk at Two Skilled Nursing Facilities with COVID-19 Outbreaks - Minnesota, April-June 2020. MMWR Morb Mortal Wkly Rep, 69(37), 1288-1295. https://doi.org/10.15585/mmwr.mm6937a3
- The Native Antigen Company. (2020, 03/24/2020). Why We Need Antigen and Antibody Tests for COVID-19. The Native Antigen Company. Retrieved 04/21/2020 from https://thenativeantigencompany.com/why-we-need-antigen-and-antibody-tests-for-covid-19/
- To, K. K. W., Yip, C. C. Y., Lai, C. Y. W., Wong, C. K. H., Ho, D. T. Y., Pang, P. K. P., Ng, A. C. K., Leung, K. H., Poon, R. W. S., Chan, K. H., Cheng, V. C. C., Hung, I. F. N., & Yuen, K. Y. (2019). Saliva as a diagnostic specimen for testing respiratory virus by a point-of-care molecular assay: a diagnostic validity study. Clin Microbiol Infect, 25(3), 372-378. https://doi.org/10.1016/j.cmi.2018.06.009 UCSD. (2020). UCSD RC SARS-CoV-2 Assay https://www.fda.gov/media/140712/download
- US. (2020, 03/27/2020). H.R.748 - CARES Act. Retrieved 05/19/2020 from https://www.congress.gov/116/bills/hr748/BILLS-116hr748enr.pdf
- Verdoni, L., Mazza, A., Gervasoni, A., Martelli, L., Ruggeri, M., Ciuffreda, M., Bonanomi, E., & D'Antiga, L. (2020). An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. https://doi.org/10.1016/s0140-6736(20)31103-x
- Villaverde, S., Domínguez-Rodríguez, S., Sabrido, G., Pérez-Jorge, C., Plata, M., Romero, M. P., Grasa, C. D., Jiménez, A. B., Heras, E., Broncano, A., Núñez, M. D. M., Illán, M., Merino, P., Soto, B., Molina-Arana, D., Bermejo, A., Mendoza, P., Gijón, M., Pérez-Moneo, B., . . . Epidemiological Study of, C.-i. C. o. t. S. S. o. P. W. G. (2021). Diagnostic Accuracy of the Panbio Severe Acute Respiratory Syndrome Coronavirus 2 Antigen Rapid Test Compared with Reverse-Transcriptase Polymerase Chain Reaction Testing of Nasopharyngeal Samples in the Pediatric Population. The Journal of pediatrics, 232, 287-289.e284. https://doi.org/10.1016/j.jpeds.2021.01.027
- Wang, F., Huang, S., Gao, R., Zhou, Y., Lai, C., Li, Z., Xian, W., Qian, X., Li, Z., Huang, Y., Tang, Q., Liu, P., Chen, R., Liu, R., Li, X., Tong, X., Zhou, X., Bai, Y., Duan, G., . . . Liu, L. (2020). Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discovery, 6(1), 83. https://doi.org/10.1038/s41421-020-00231-4
- Wang, R., Qian, C., Pang, Y., Li, M., Yang, Y., Ma, H., Zhao, M., Qian, F., Yu, H., Liu, Z., Ni, T., Zheng, Y., & Wang, Y. (2020). opvCRISPR: One-pot visual RT-LAMP-CRISPR platform for SARS-cov-2 detection. Biosensors and Bioelectronics, 172, 112766. https://doi.org/https://doi.org/10.1016/j.bios.
2020.112766 - WHO. (2020a, 09/11/20). Diagnostic testing for SARS-CoV-2. Retrieved 11/08/20 from https://www.who.int/publications/i/item/diagnostic-testing-for-sars-cov-2
- WHO. (2020b, 04/24/2020). "Immunity passports" in the context of COVID-19. World Health Organization. Retrieved 04/25/2020 from https://www.who.int/news-room/commentaries/detail/immunity-passports-in-the-context-of-covid-19
- WHO. (2020c, 05/15/2020). Multisystem inflammatory syndrome in children and adolescents with COVID-19. World Health Organization. Retrieved 05/18/2020 from https://www.who.int/publications-detail/multisystem-inflammatory-syndrome-in-children-and-adolescents-with-covid-19
- WHO. (2021a, October 6). Antigen-detection in the diagnosis of SARS-CoV-2 infection. World Health Organization. Retrieved 11/08/2020 from https://www.who.int/publications/i/item/antigen-detection-in-the-diagnosis-of-sars-cov-2infection-using-rapid-immunoassays
- WHO. (2021b, November 2021). COVID-19 Clinical management: living guidance. World Health Organization. Retrieved April 19 from https://apps.who.int/iris/bitstream/handle/10665/349321/WHO-2019-nCoV-clinical-2021.2-eng.pdf
- WHO. (2021c). COVID-19 natural immunity. file:///C:/Users/AHCS8330/Downloads/WHO-2019-nCoV-Sci-Brief-Natural-immunity-2021.1-eng.pdf
- WHO. (2022). Use of SARS-CoV-2 antigen-detection rapid diagnostic tests for COVID-19 self-testing. https://www.who.int/publications/i/item/WHO-2019-nCoV-Ag-RDTs-Self_testing-2022.1
- WHO. (2023, November 11). Coronavirus disease (COVID-19) Pandemic. World Health Organization. https://www.who.int/emergencies/diseases/novel-coronavirus-2019
- WHO. (2024a, 2022). Middle East respiratory syndrome coronavirus (MERS-CoV). World Health Organization. Retrieved 08/19/2020 from https://www.who.int/emergencies/mers-cov/en/
- WHO. (2024b). SARS (Severe Acute Respiratory Syndrome). World Health Organization. Retrieved 04/19/2022 from https://www.who.int/ith/diseases/sars/en/
- Woof, J. M., & Kerr, M. A. (2006). The function of immunoglobulin A in immunity. The Journal of Pathology, 208(2), 270-282. https://doi.org/10.1002/path.1877
- Wu, F., Liu, M., Wang, A., Lu, L., Wang, Q., Gu, C., Chen, J., Wu, Y., Xia, S., Ling, Y., Zhang, Y., Xun, J., Zhang, R., Xie, Y., Jiang, S., Zhu, T., Lu, H., Wen, Y., & Huang, J. (2020). Evaluating the Association of Clinical Characteristics With Neutralizing Antibody Levels in Patients Who Have Recovered From Mild COVID-19 in Shanghai, China. JAMA Intern Med. https://doi.org/10.1001/jamainternmed.2020.4616
- Wulff, N. H., Tzatzaris, M., & Young, P. J. (2012). Monte Carlo simulation of the Spearman-Kaerber TCID50. J Clin Bioinforma, 2(1), 5. https://doi.org/10.1186/2043-9113-2-5
- Xiao, D. A. T., Gao, D. C., & Zhang, D. S. (2020). Profile of Specific Antibodies to SARS-CoV-2: The First Report. J Infect. https://doi.org/10.1016/j.jinf.2020.03.012
- Yang, X., Yu, Y., Xu, J., Shu, H., Xia, J., Liu, H., Wu, Y., Zhang, L., Yu, Z., Fang, M., Yu, T., Wang, Y., Pan, S., Zou, X., Yuan, S., & Shang, Y. (2020). Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med, 8(5), 475-481. https://doi.org/10.1016/s2213-2600(20)30079-5
- Yau, F., Ferreira, R., Kamali, R., Bird, P. W., Halliwell, R., Patel, H., Nicoara, D. C., Woltmann, G., & Tang, J. W. (2021). Clinical utility of a rapid 'on-demand' laboratory-based SARS-CoV-2 diagnostic testing service in an acute hospital setting admitting COVID-19 patients. Clin Infect Pract, 12, 100086. https://doi.org/10.1016/j.clinpr.
2021.100086 - Yelin, I., Aharony, N., Shaer Tamar, E., Argoetti, A., Messer, E., Berenbaum, D., Shafran, E., Kuzli, A., Gandali, N., Shkedi, O., Hashimshony, T., Mandel-Gutfreund, Y., Halberthal, M., Geffen, Y., Szwarcwort-Cohen, M., & Kishony, R. (2020). Evaluation of COVID-19 RT-qPCR test in multi-sample pools. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa531
- Zhang, Y. V., Wiencek, J., Meng, Q. H., Theel, E. S., Babic, N., Sepiashvili, L., Pecora, N. D., Slev, P., Cameron, A., Konforte, D., & the, A. C. S. T. T. F. (2021). AACC Practical Recommendations for Implementing and Interpreting SARS-CoV-2 EUA and LDT Serologic Testing in Clinical Laboratories. Clinical Chemistry. https://doi.org/10.1093/clinchem/hvab051
- Zhao, J., Yuan, Q., Wang, H., Liu, W., Liao, X., Su, Y., Wang, X., Yuan, J., Li, T., Li, J., Qian, S., Hong, C., Wang, F., Liu, Y., Wang, Z., He, Q., Li, Z., He, B., Zhang, T., . . . Zhang, Z. (2020). Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clinical Infectious Diseases. https://doi.org/10.1093/cid/ciaa344
Coding Section
| CPT |
Code Description |
| 86328 |
Immunoassay for infectious agent antibody(ies), qualitative or semiquantitative, single step method (e.g., reagent strip); severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronavirus disease [COVID-19]) |
| 86408 |
Neutralizing antibody, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronavirus disease [COVID19]); screen |
| 86409 |
Neutralizing antibody, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronavirus disease [COVID19]); titer |
| 86413 |
Severe acute respiratory syndrome coronavirus 2 (SARSCoV-2) (Coronavirus disease [COVID-19]) antibody, quantitative |
| 86769 |
Antibody; severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronavirus disease [COVID-19]) |
| 87426 |
Infectious agent antigen detection by immunoassay technique, (e.g., enzyme immunoassay [EIA], enzyme-linked immunosorbent assay [ELISA], immunochemiluminometric assay [IMCA]) qualitative or semiquantitative, multiple-step |
| 87635 |
Infectious agent detection by nucleic acid (DNA or RNA); severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronavirus disease [COVID-19]), amplified probe technique |
| 87798 |
Infectious agent detection by nucleic acid (DNA or RNA), not otherwise specified; amplified probe technique, each organism |
| 87811 |
Infectious agent antigen detection by immunoassay with direct optical (i.e., visual) observation; severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronavirus disease [COVID-19]) |
| 87913 | Infectious agent genotype analysis by nucleic acid (DNA or RNA); severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (coronavirus disease [COVID-19]), mutation identification in targeted region(s) |
| 0224U |
Antibody, severe acute respiratory syndrome |
| 0226U |
Surrogate viral neutralization test (sVNT), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronavirus disease [COVID-19]), ELISA, plasma, serum |
| 0408U |
Infectious agent antigen detection by bulk acoustic wave biosensor immunoassay, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (coronavirus disease [COVID-19]) Proprietary test: Omnia™ SARS-CoV-2 Antigen Test Lab/Manufacturer: Qorvo Biotechnologies |
| U0001 |
CDC Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel |
| U0002 |
Non-CDC laboratory test for 2019-nCoV (COVID-19), any method |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2023 Forward