
Genetic Testing for Alpha- and Beta-Thalassemia - CAM 297
Description
Alpha-thalassemia is characterized by an impaired production of the alpha globin chains of hemoglobin, leading to a relative excess of gamma globin chains (fetus and newborn), or excess beta globin chains (children and adults) mainly due to deletion or mutation of the alpha globin genes. There are four alpha-thalassemia syndromes, reflecting the loss of function of one, two, three, or all four of these alpha chain genes varying in severity from non-symptomatic to incompatibility with extrauterine life.1,2
Beta-thalassemia is similarly characterized by impaired production of hemoglobin components but affects the beta chains instead of the alpha chains. This creates excess alpha globin chains, leading to hemolytic anemia, impaired iron handling, and other clinical symptoms.3
When pursuing genetic testing for alpha- or beta-thalassemia, genetic counseling is strongly recommended.
Terms such as male and female/woman are used when necessary to refer to sex assigned at birth.
For guidance on prenatal screening and preconception screening for alpha- or beta-thalassemia, please see CAM 358 Prenatal Screening (Genetic).
Background
Hemoglobin, which is the major oxygen carrying protein molecule of red blood cells, consists of 2 α-globin chains and 2 β-globin chains. Alpha-thalassemia refers to a group of syndromes that arise from deficient production of α-globin chains. Deficient α-globin production leads to an excess of β-globin chains, which results in anemia by a number of mechanisms1:
-
Ineffective erythropoiesis in the bone marrow
-
Production of nonfunctional hemoglobin molecules
-
Shortened survival of RBCs [red blood cells] due to intravascular hemolysis and increased uptake of the abnormal RBCs by the liver and spleen
The physiologic basis of α-thalassemia is a genetic defect in the genes coding for α-globin production. Each individual carries 4 genes that code for α-globin (2 copies each of HBA1 and HBA2, located on chromosome 16), with the wild genotype (normal) being aa/aa. Genetic mutations may occur in any or all of these 4 α-globin genes. The number of genetic mutations determines the phenotype and severity of the α-thalassemia syndromes. The different syndromes are classified as follows:
- Silent carrier (α-thalassemia minima). This arises from 1 of 4 abnormal alpha genes (aa/a-), and is a silent carrier state. A small amount of abnormal hemoglobin can be detected in the peripheral blood, and there may be mild hypochromia and microcytosis present, but there is no anemia or other clinical manifestations.
- Thalassemia trait (α-thalassemia minor). This is also called α-thalassemia trait and arises from the loss of 2 α-globin genes, resulting on 1 of 2 genotypes (aa/--, or a-/a-). There is a mild anemia present, and red blood cells are hypochromic and microcytic. Clinical symptoms are usually absent and in most cases, the Hg electrophoresis is normal.
- Hemoglobin H disease (α-thalassemia intermedia). This syndrome results from 3 abnormal α-globin genes (a-/--), resulting in a moderate to severe anemia. In HgH disease, there is an imbalance in α- and β-globin gene chain synthesis, resulting in the precipitation of excess β chains into the characteristic hemoglobin H, or β-tetramer. This condition has marked phenotypic variability, but most individuals have mild disease and live a normal life without medical intervention.2
A minority of individuals may develop clinical symptoms of chronic hemolytic anemia. These include neonatal jaundice, hepatosplenomegaly, hyperbilirubinemia, leg ulcers, and premature development of biliary tract disease. Splenomegaly can lead to the need for splenectomy, and transfusion support may be required by the third to fourth decade of life. It has been estimated that approximately 25% of patients with HgH disease will require transfusion support during their lifetime.3 In addition, increased iron deposition can lead to premature damage to the liver and heart. Inappropriate iron therapy and oxidant drugs should be avoided in patients with HgH disease.
There is an association between genotype and phenotype among patients with HgH disease. Individuals with a nondeletion mutation typically have an earlier presentation, more severe anemia, jaundice, and bone changes, and more frequently require transfusions.4
- Hemoglobin Bart syndrome (α-thalassemia major). This syndrome results from mutations in all 4 α-globin genes (--/--), resulting in absent production of α-globin chains. This condition causes hydrops fetalis, which often leads to intrauterine death, or death shortly after birth. There are also increased complications of pregnancy for a woman carrying a fetus with hydrops fetalis. These include hypertension, preeclampsia, antepartum hemorrhage, renal failure, premature labor, and abruption placenta.3
Alpha-thalassemia is a common genetic disorder, affecting approximately 5% of the world’s population.3 The frequency of mutations is highly dependent on ethnicity, with the highest rates seen in Asians, and much lower rates in Northern Europeans. The carrier rate is estimated to be 1 in 20 in Southeast Asians, 1 in 30 for Africans, and between 1 in 30 and 1 in 50 for individuals of Mediterranean ancestry. In contrast, for individuals of northern European ancestry, the carrier rate is less than 1 in 1,000.
Genetic Testing
A number of different types of genetic abnormalities are associated with α-thalassemia. More than 100 different genetic mutations have been described. Deletion of 1 or more of the α-globin chains is the most common genetic defect. This is the type of genetic defect found in approximately 90% of cases.5 Large genetic rearrangements can also occur from defects in crossover and/or recombination of genetic material during reproduction. Point mutations in 1 or more of the α genes can occur that impair transcription and/or translation of the α-globin chains.
Testing is commercially available through several genetic labs. Targeted mutation analysis for known α-globin gene mutations can be performed by polymerase chain reaction (PCR).4,5 PCR can also be used to identify large deletions or duplications. Newer testing methods have been developed to facilitate identification of α-thalassemia mutations, such as multiplex amplification methods and real-time PCR analysis.6-8 In patients with suspected α-thalassemia and a negative PCR test for genetic deletions, direct sequence analysis of the α-globin locus is generally performed to detect point mutations.5
Regulatory Status
Genetic testing for alpha thalassemia is available as a laboratory-developed service, subject only to the general laboratory operational regulation under the Clinical Laboratory Improvement Amendments (CLIA) of 1988. Laboratories performing clinical tests must be certified for high-complexity testing under CLIA. The U.S. Food and Drug Administration (FDA) has not regulated these tests to date.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- Genetic testing to confirm an alpha- or beta-thalassemia diagnosis is considered MEDICALLY NECESSARY in any of the following situations:
- For individuals for whom one parent is a known carrier of alpha- or beta-thalassemia
- For individuals for whom other testing to diagnose the cause of microcytic anemia has been inconclusive
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- For all other situations not described above, genetic testing for alpha- or beta-thalassemia is considered NOT MEDICALLY NECESSARY.
Table of Terminology
| Term |
Definition |
| ACMG |
American College of Medical Genetics and Genomics |
| ACOG |
American College of Obstetrics and Gynecology |
| APHL |
The Association of Public Health Laboratories |
| BCL11A |
B-cell CLL/lymphoma 11a |
| BSH |
British Society for Haematology |
| CCMG |
Canadian College of Medical Geneticists |
| CLIA ’88 |
Clinical Laboratory Improvement Amendments Of 1988 |
| CMS |
Centers for Medicare & Medicaid |
| CRISPR/Cas9 |
Clustered Regularly Interspaced Short Palindromic Repeated/CRISPR-associated protein 9 |
| DNA |
Deoxyribonucleic acid |
| FDA |
Food And Drug Administration |
| Hb |
Hemoglobin |
| HBA1 |
Alpha Globin 1 |
| HBA2 |
Alpha Globin 2 |
| HBB |
Hemoglobin, subunit beta |
| HbF |
Hemoglobin F |
| HE |
Electrophoresis |
| HHPLC |
Hb high performance liquid chromatography |
| HLA |
Human leukocyte antigens |
| LDTs |
Laboratory-developed tests |
| MCH |
Mean cell hemoglobin |
| MCV |
Mean corpuscular volume |
| MDA |
Multiple displacement amplification |
| NGS |
Next-generation sequencing |
| NHS |
National Health Service |
| PCR |
Polymerase chain reaction |
| PGT |
Preimplantation genetic testing |
| PHE |
Public Health England |
| SCT |
Sickle cell and thalassemia |
| SNP |
Single nucleotide polymorphism |
| SOGC |
Society of Obstetricians and Gynaecologists of Canada |
| β+ |
Reduced expression |
| β0 |
Absent expression |
Rationale
Thalassemias result from deficiencies in hemoglobin biosynthesis due to mutations in or near the two globin gene clusters which encode the globin polypeptide subunits of hemoglobin.2 Normal hemoglobin is a heterotetramer of two alpha globin chains and two beta globin chains (hemoglobin A) or two gamma globin chains (hemoglobin F). Well over 100 mutations have been documented to affect the biosynthesis or post-translational stability of the globin subunits needed for successful production of the large amounts of Hb needed for normal red cell homeostasis. Globin chain synthesis is very tightly controlled, such that the ratio of production of alpha to non-alpha chains is almost exactly 1:1.4
Alpha-thalassemia refers to thalassemias that result from impaired or absent production of alpha globin, leading to a relative excess of gamma globin (fetus and newborn), or excess beta globin (children and adults). Excess beta globin chains can form soluble homotetramers, but they are nonfunctional and unstable. This may lead to increased hemolysis and a variety of clinical manifestations, such as anemia, thrombosis, and skeletal changes. A diagnosis of alpha-thalassemia is often confirmed by genetic testing, as assessment of the hemoglobin gene is inexpensive and convenient.4
The clinical severity is directly attributable to the net deficit of alpha globin synthesis but is complicated by the number of alpha globin genes affected, which of the two alpha globin loci is affected, and the degree to which the mutation blocks gene expression. In addition, combinations of defects in both alpha and beta globulins can balance each other out. Thus, understanding the broad spectrum of clinical severity in alpha-thalassemia requires a detailed knowledge of the underlying genetic defect and the impact of these defects on the overall levels and balance of globin chain synthesis.3
The majority of cases of alpha-thalassemia are attributable to deletion of alpha globin alleles, especially in Asia and Africa.5 However, more detailed analysis of globin gene sequences suggests that some fairly common forms of alpha-thalassemia that appear to arise from a deletion of one copy of an alpha globin gene are actually due to unequal crossover and recombination events that fuse the two alpha globin genes together into one.4 Additionally, non-deletion alleles are also common, especially in the Mediterranean area, which contain mutations producing highly unstable alpha globin variants unable to produce intact hemoglobin.2 Current research continues to identify novel mutations and improve thalassemia screening.6
Beta-thalassemia is similar to alpha-thalassemia, with the beta chains of hemoglobin affected instead of the alpha chains. However, excess alpha globin chains do not form soluble homotetramers, causing them to aggregate when they accumulate in erythroid precursors. This causes clinical symptoms to be more severe, although the symptoms themselves are similar to alpha-thalassemia (anemia, iron overload, and so on).2,3 There are two beta globin genes compared to four for the alpha chain. As with alpha-thalassemia, the severity of clinical presentation depends on the genotype of the beta globin genes (i.e. the ratio of beta to alpha globin chains). Mutations may result in a reduced expression (β+) or absent expression (β0). β0 phenotypes are generally transfusion-dependent as they produce very little (if any) adult hemoglobin.3
Due to the frequency of thalassemias worldwide, carrier screening may be useful, particularly in areas such as Southeast Asia, Africa, and the Indian subcontinent. Both primary thalassemias are autosomal recessive genetic disorders so parents who are heterozygous carriers would have a 25% chance to have an affected child despite being asymptomatic themselves. Identification of an affected fetus could alter decisions during the pregnancy.7
Below is a table summarizing the clinical genotypes and phenotypes of both thalassemia syndromes.2,8,9
Analytical Validity
He, et al. (2017) examined a next-generation sequencing (NGS) panel’s utility for thalassemia screening in Southwestern China. A total of 951 individuals were tested, and the NGS screen found 471 carriers (49.5%) of thalassemia. In comparison, traditional methods (defined as “red cell indexes and hemoglobin electrophoresis, then DNA sequencing”) identified only 209 carriers (22%) of thalassemia, missing 217 alpha-thalassemia carriers and 47 beta-thalassemia carriers.10
In a separate study by Zhang, et al. (2019), because of studying 3,973 subjects that underwent hematological examinations and additional NGS and Gap-PCR due to being suspected thalassemia carriers, the researchers found that “approximately 2.88% thalassemia carriers would be missed by traditional genetic analysis. In addition, four novel thalassemia mutations and one novel abnormal hemoglobin mutation were identified.” This research further corroborated the increased effectiveness of using NGS in screening thalassemia in an area of high disease prevalence.11
Shook, et al. (2020) evaluated the accuracy of a specific pattern in hemoglobin separation tests. The authors desired to find if an “FSA” pattern corresponded to a final diagnosis of the sickle cell trait (HbAS), or a final diagnosis of sickle beta-thalassemia (HbSβ+). Traditionally, the FSA pattern has indicated a diagnosis of HbSβ+; however, the authors hypothesized that the FSA pattern truly indicates a diagnosis of HbAS instead. A total of 31 newborns with an initial screening result of the FSA pattern (a suspected diagnosis of HbSβ+) were included. There were 30 newborns that underwent protein-base confirmatory testing and 17 underwent confirmatory genetic testing. Of the newborns undergoing protein confirmatory testing, 23 had an “FSA” pattern, establishing a diagnosis of HbAS. Of the eight remaining newborns with an FSA pattern, seven underwent genetic testing which identified HbAS as well. Genetic testing also confirmed positive HbAS results in ten newborns that tested initially positive by protein testing. The authors concluded that genetic testing had utility in newborn screening for hemoglobinopathies.12
Chen, et al. (2021) established an effective NGS protocol for four-factor preimplantation genetic testing (PGT) to diagnose α- and β-thalassemia. Three couples, in whom both partners were α- and β-double thalassemia carriers, underwent PGT and a total of 35 biopsied trophectoderm samples underwent multiple displacement amplification (MDA). Using NGS-based single nucleotide polymorphism (SNP) haplotyping, these samples were analyzed. A total of “51.5% (17/33) of the embryos were diagnosed as unaffected non-carriers or carriers. Of the 17 unaffected embryos, nine (52.9%) were tested further and identified as euploid via NGS-based aneuploid screening, in which five had HLA types matching affected children.” The authors conclude that NGS-SNP was effective in performing PGT for multipurpose detection.13
Clinical Utility and Validity
Nosheen, et al. (2015) evaluated a preliminary screening program for beta-thalassemia. The screening program focused on families of beta-thalassemia major children. There were 98 samples taken, and 57 were found to have a beta-thalassemia trait with elevated hemoglobin alpha 2. The mean hemoglobin alpha 2 level of the carriers was 5.2±0.56% compared to 2.34±0.57% in normal subjects. The authors suggested that screening programs and counseling for carriers could decrease incidence of beta-thalassemia major.14
Satirapod, et al. (2019) evaluated the clinical outcomes of using PGT in couples at risk of passing on beta-thalassemia. Two components of PGT were used, PGT for monogenic disease (used for diagnosis) and PGT for aneuploidy (intended to identify chromosomal aberrations) A total of 15 couples were included and a total of 106 embryos were tested. After preimplantation testing, 12 of 15 individuals were able to obtain satisfactory genetic testing results (defined as non-disease affected embryos without chromosomal aberration and transfer within first two cycles). Of these, nine individuals had successful implantations and eight individuals had successful pregnancies with live births (deemed a 53.33% success rate). PGT assessment of genetic status was confirmed by pre- and post-natal genetic testing. Overall, the authors concluded that combined PGT-A and PGT-M was a useful technology to prevent beta-thalassemia in the offspring of recessive carriers. To increase the diagnostic efficiency of PGT-M, MDA may be utilized as the first step. This conclusion was drawn by Fu, et al. (2019), who found in a retrospective cohort study, that from 2,315 embryos tested, MDA yielded a 96.99% diagnostic efficiency, versus a PCR group, which only yielded 88.15%. MDA also enabled statistically significantly more embryos to be available for transfer as well when compared to the PCR group (74.28% vs 64.28%, respectively, P < 0.001).16
Chen, et al. (2020) also conducted a similar study that evaluated “the efficacy of preimplantation genetic testing (PGT) for α- and β-double thalassemia combined with aneuploidy screening using next-generation sequencing (NGS).” From 12 couples that each carried both α- and β- thalassemia mutations, the researchers were able to facilitate 11 healthy live births from examining 112 embryos. This NGS-based SNP haplotyping was demonstrated to “reduce misdiagnosis by linkage analyses with multiple SNP loci” and increase the number of diagnosis results, including those from detecting aneuploidy and identified mutation sites, in a single PGT cycle. It was also found that NGS-based SNP haplotyping could be performed “through directly detecting mutation sites with NGS and using affected embryos or gametes as probands.” This procedure benefits in eliminating multiple biopsies as well.17
Dan, et al. (2023) conducted a literature mini-review of the current state of beta-thalessemia management. Currently, beta-thalessemia requires lifelong management strategies that include regular blood transfusions and iron chelation therapy for severe cases. The review discussed the limitations of current therapies and advocates for continued research into hematopoietic stem cell transplantation and gene therapy as new treatment approaches. Several novel therapeutic methods are being explored for clinical utility. For example, Luspatercept is a recently FDA-approved therapy that works by inhibiting the Smad2/3 signaling pathway, which is involved in erythropoiesis (the process of producing red blood cells). By modifying this pathway, Luspatercept helps to reduce the ineffective erythropoiesis that is a hallmark of beta-thalessemia. It is particularly useful for patients who are transfusion-dependent and has purported promise in improving iron balance and reducing the frequency of blood transfusions; however, the cost of Luspatercept is $170,000 annually per patient. Hydroxyurea is a drug that is well-known in the treatment of blood disorders, such as sickle cell disease, but could also have potential benefits in treating beta-thalessemia. Gene therapy focuses on strategies to increase the production of gamma globin chains, thereby increasing HbF levels. This approach could potentially correct the underlying genetic defects causing thalassemia. Gene therapy techniques include: (1) CRISPR/Cas9 gene editing (involves editing the BCL11A gene, a key regulator of hemoglobin production, to enhance HbF synthesis) and (2) Lentiviral Gene Transfer (therapies that involve a lentiviral vector to insert a functional copy of the beta-globin gene into the patients’ hematopoietic stem cells). One example of lentiviral gene transfer is Zynteglo gene therapy, the first FDA-approved genetic treatment for beta-thalessemia.18
American College of Medical Genetics and Genomics (ACMG)
In 2021, ACMG released an updated guideline for screening for autosomal recessive and X-linked conditions during pregnancy and preconception. Their practice resource aims to recommend “a consistent and equitable approach for offering carrier screening to all individuals during pregnancy and preconception” and replaces any earlier ACMG position statements on prenatal/preconception expanded carrier screening.
The ACMG provides carrier screening recommendations during pregnancy that are split into three tiers. Tier 1 includes recommended screenings for all couples considering pregnancy or pregnant women. The tier 1 recommendations include disorders that have significant health impacts, for which prenatal diagnosis and potential interventions might be available; this tier includes screenings relevant to this policy, such as sickle cell disease, alpha-thalassemia, and beta-thalassemia. Tier 2 includes additional screenings that may be offered based on the family history or ancestry that suggest higher risk of specific genetic conditions. In some cases, expanded carrier screening can be considered, which may test for less common hemoglobinopathies. Tier 3 screenings are “optional” and can be considered based on individual or family history factors. These screenings may include rare genetic disorders or less common variants of hemoglobinopathies. The ACMG provides the following specific recommendations:
- “Carrier screening enables those screened to consider their reproductive risks, reproductive options, and to make informed decisions.”
- “The phrase ‘expanded carrier screening’ be replaced by ‘carrier screening’.”
- “Adopting a more precise tiered system based on carrier frequency:
- Tier 4: <1/200 carrier frequency (includes Tier 3) genes/condition will vary by lab
- Tier 3: ≥ 1/200 carrier frequency (includes Tier 2) includes X-linked conditions
- Tier 2: ≥1/100 carrier frequency (includes Tier 1)
- Tier 1: CF [Cystic Fibrosis] + SMA [spinal muscular atrophy] + Risk Based Screening”
- “Tier 1 screening conveys the recommendations previously adopted by ACMG and ACOG” and “adopts an ethnic and population neutral approach when screening for cystic fibrosis and spinal muscular atrophy. Beyond these two conditions, additional carrier screening is determined after risk assessment, which incorporates personal medical and family history as well as laboratory and imaging information where appropriate”
- “Tier 2 carrier screening stems from an ACOG recommendation for conditions that have a severe or moderate phenotype and a carrier frequency of at least 1/100.” However, “data demonstrate that carrier screening for two common conditions using a carrier frequency threshold of 1/100 may not be equitable across diverse populations. Others have shown that limiting the carrier frequency to ≥1/100 creates missed opportunities to identify couples at risk for serious conditions.”
- “We define Tier 3 screening as carrier screening for conditions with a carrier frequency ≥1/200 . . . Tier 2 and Tier 3 screening prioritize carrier frequency as a way to think about conditions most appropriate for screening in the general population. However, when ACOG proposed this level, they did not specify whether it was thinking about carrier frequency in terms of the global population or subpopulations. We use ‘carrier frequency’ to mean in any ethnic group with reasonable representation in the United States.”
- “Tier 4 includes genes less common than those in Tier 3 and can identify additional at-risk couples. Tier 4 has no lower limit carrier screening frequency and can greatly extend the number of conditions screened . . . the clinical validity at this level of carrier screening may be less compelling, therefore we suggest reserving this level of screening for consanguineous pregnancies (second cousins or closer) and in couples where family or medical history suggests Tier 4 screening might be beneficial . . . Importantly, patients should understand that their chance of being a carrier for one or more conditions increases as the number of conditions screened is increased.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening.”
- Tier 4 screening should be considered:
- When a pregnancy stems from a known or possible consanguineous relationship (second cousins or closer);
- When a family or personal medical history warrants.
- ACMG does NOT recommend:
- Offering Tier 1 and/or Tier 2 screening, because these do not provide equitable evaluation of all racial/ethnic groups.
- Routine offering of Tier 4 panels.
- “Carrier screening paradigms should be ethnic and population neutral and more inclusive of diverse populations to promote equity and inclusion.”
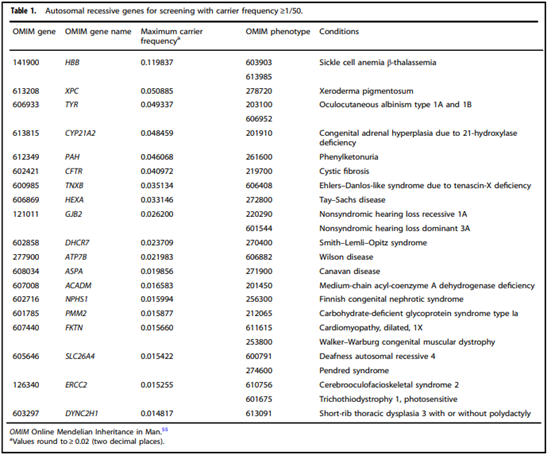
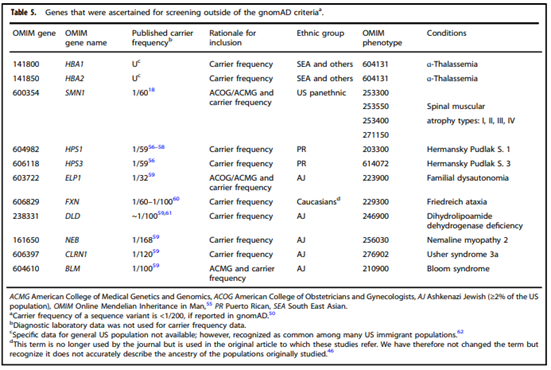
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening for autosomal recessive [Table 1 & 5] . . . conditions.”
- “Reproductive partners of pregnant patients and those planning a pregnancy may be offered Tier 3 carrier screening for autosomal recessive conditions [Table 1 & 5] when carrier screening is performed simultaneously with their partner.”
- “When Tier 1 or Tier 2 carrier screening was performed in a prior pregnancy, Tier 3 screening should be offered.”19
Canadian College of Medical Geneticists (CCMG) and Society of Obstetricians and Gynaecologists of Canada (SOGC)
The CCMG and SOGC published a joint guideline titled “Carrier Screening for Thalassemia and Hemoglobinopathies in Canada” in 2008. Their recommendations addressing thalassemia’s/hemoglobinopathies are listed below:
- “Carrier screening for thalassemia and hemoglobinopathies should be offered to a woman if she and/or her partner are identified as belonging to an ethnic population whose members are at higher risk of being carriers. Ideally, this screening should be done pre-conceptionally or as early as possible in the pregnancy. (II-2A)
- Screening should consist of a complete blood count, as well as hemoglobin electrophoresis or hemoglobin high performance liquid chromatography. This investigation should include quantitation of HbA2 and HbF. In addition, if there is microcytosis (mean cellular volume < 80 fL) and/or hypochromia (mean cellular hemoglobin < 27 pg) in the presence of a normal hemoglobin electrophoresis or high-performance liquid chromatography the patient should be investigated with a brilliant cresyl blue stained blood smear to identify H bodies. A serum ferritin (to exclude iron deficiency anemia) should be performed simultaneously. (III-A)
- If a woman’s initial screening is abnormal (e.g., showing microcytosis or hypochromia with or without an elevated HbA2, or a variant Hb on electrophoresis or high-performance liquid chromatography) then screening of the partner should be performed. This would include a complete blood count as well as hemoglobin electrophoresis or HPLC, HbA2 and HbF quantitation, and H body staining. (III-A)
- If both partners are found to be carriers of thalassemia or an Hb variant, or of a combination of thalassemia and a hemoglobin variant, they should be referred for genetic counselling. Ideally, this should be prior to conception, or as early as possible in the pregnancy. Additional molecular studies may be required to clarify the carrier status of the parents and thus the risk to the fetus. (II-3A)
- Prenatal diagnosis should be offered to the pregnant woman/couple at risk for having a fetus affected with a clinically significant thalassemia or hemoglobinopathy. Prenatal diagnosis should be performed with the patient’s informed consent. If prenatal diagnosis is declined, testing of the child should be done to allow early diagnosis and referral to a pediatric hematology centre, if indicated. (II-3A)
- Prenatal diagnosis by DNA analysis can be performed using cells obtained by chorionic villus sampling or amniocentesis. Alternatively, for those who decline invasive testing and are at risk of hemoglobin Bart’s hydrops fetalis (four-gene deletion α-thalassemia), serial detailed fetal ultrasound for assessment of the fetal cardiothoracic ratio (normal < 0.5) should be done in a centre that has experience conducting these assessments for early identification of an affected fetus. If an abnormality is detected, a referral to a tertiary care centre is recommended for further assessment and counselling. Confirmatory studies by DNA analysis of amniocytes should be done if a termination of pregnancy is being considered. (II-3A)
- The finding of hydrops fetalis on ultrasound in the second or third trimester in [individuals] with an ethnic background that has an increased risk of α-thalassemia should prompt immediate investigation of the pregnant patient and her partner to determine their carrier status for α-thalassemia.
In a SOGC clinical practice guideline for Investigation and Management of Non-immune Fetal Hydrops, the society recommends “investigation for maternal-fetal infections and alpha-thalassemia in women at risk because of their ethnicity should be performed in all cases of unexplained fetal hydrops.”21
The Thalassemia Longitudinal Cohort
The report on the Thalassemia Longitudinal Cohort recommends: “Obtaining genotyping to confirm the diagnosis and HLA typing for transplant evaluation for all patients who require chronic transfusion is strongly recommended. For pediatric patients, annual comprehensive follow up should include assessment of the availability of a related donor as well as a recommendation to bank cord blood and obtain HLA typing on all subsequently born full siblings.”22
American College of Obstetrics and Gynecology (ACOG)
The ACOG Committee Opinion #691 (“Carrier Screening for Genetic Conditions”) states that: “Couples at risk of having a child with a hemoglobinopathy may benefit from genetic counseling to review their risk, the natural history of these disorders, prospects for treatment and cure, availability of prenatal genetic testing, and reproductive options. Prenatal diagnostic testing for the mutation responsible for sickle cell disease is widely available. Testing for α-thalassemia and β-thalassemia is possible if the mutations and deletions have been previously identified in both parents. These DNA-based tests can be performed using chorionic villi obtained by chorionic villus sampling or using cultured amniotic fluid cells obtained by amniocentesis. For some couples, preimplantation genetic diagnosis in combination with in vitro fertilization may be a desirable alternative to avoid termination of an affected pregnancy. Preimplantation genetic diagnosis has been successfully performed for sickle cell disease and most types of β-thalassemia.”23 This was reaffirmed in 2023.
In 2022, ACOG put out a new practice bulletin regarding hemoglobinopathies in pregnancy.24 This was also reaffirmed in September, 2024.
- The ACOG “recommends offering universal hemoglobinopathy testing to persons planning pregnancy or at the initial prenatal visit if no prior testing results are available for interpretation.”
- “Hemoglobinopathy testing may be performed using hemoglobin electrophoresis or molecular genetic testing (eg, expanded carrier screening that includes sickle cell disease [SCD] and other hemoglobinopathies).”
- “The use of noninvasive prenatal diagnosis for SCD with cell-free fetal DNA is still experimental and currently not recommended.”24
National Health Service (NHS)
The NHS released standards for antenatal laboratories working with the NHS sickle cell and thalassaemia (SCT) screening program.25 The referral guidelines for antenatal screening specimens are as follows:


The Association of Public Health Laboratories (APHL)
The APHL states that “molecular testing can be added to resolve cases when the newborn has been transfused with packed red blood cells. Since the newborn’s phenotype is masked by the donor, DNA testing can be used to identify any abnormal hemoglobins.”26
Public Health England (PHE)
The PHE highlights the importance of antenatal screening. If the baby’s mother is identified as a carrier, the biological father should also be tested. Both prenatal diagnosis and genetic counseling are recommended by the PHE.27
British Society for Haematology (BSH)
The BSH provides the following recommendations:
- “Antenatal screening/testing of pregnant [individuals] should be carried out according to the guidelines of the NHS Sickle Cell and Thalassaemia Screening programme.
- Laboratories performing antenatal screening should utilize methods capable of detecting significant variants and be capable of quantitating haemoglobins A2 and F at the cut‐off points required by the national antenatal screening program.”28
Genetic counseling is also permitted for prospective parents.
The Thalassemia International Foundation (TIF)
The TIF provided recommendations for the management of transfusion-dependent thalassemia. The following recommendations were made:29
- “Molecular genetic testing is available in clinical laboratories and may be useful for predicting the clinical phenotype in some cases as well as enabling presymptomatic diagnosis of at-risk family members and prenatal diagnosis.
- Molecular analysis is not required to confirm the diagnosis of a β carrier, but it is necessary to confirm the α thalassemia carrier status (grade A)
- Since the prevalent pathogenic variants of the β globin gene are limited in each at-risk population, a PCR method designed to detect the common specific mutation simultaneously should be used initially (grade B)
- β globin gene sequence analysis may be considered first if the affected individual is not of an ancestry at high risk or if targeted analysis reveals only one or no pathogenic variant (grade B)
- α thalassemias are mainly due to deletions of different length and they can be detected preferentially by reverse dot blot and Gap-PCR (grade B)
- Methods that may be used to detect rare or unknown deletions include: Southern blotting (now fallen into abeyance), quantitative PCR, long-range PCR and, above all, MLPA (grade B).”29
References
- Martin A, Thompson AA. Thalassemias. Pediatric clinics of North America. Dec 2013;60(6):1383-91. doi:10.1016/j.pcl.2013.08.008
- Benz Jr EJ, Angelucci E. Clinical manifestations and diagnosis of the thalassemias. Updated November 21, 2024. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-the-thalassemias
- Benz Jr EJ. Pathophysiology of thalassemia. Updated June 26, 2024. https://www.uptodate.com/contents/pathophysiology-of-thalassemia
- Benz Jr EJ. Molecular pathology of the thalassemic syndromes. Updated September 11, 2024. https://www.uptodate.com/contents/molecular-genetics-of-the-thalassemia-syndromes
- Steinberg MH. Management of sickle cell disease. The New England journal of medicine. Apr 01 1999;340(13):1021-30. doi:10.1056/NEJM199904013401307
- He S, Li J, Li DM, et al. Molecular characterization of alpha- and beta-thalassemia in the Yulin region of Southern China. Gene. May 20 2018;655:61-64. doi:10.1016/j.gene.2018.02.058
- Yates A. Hemoglobinopathy: Screening and counseling in the reproductive setting and fetal diagnosis. Updated June 2, 2025. https://www.uptodate.com/contents/hemoglobinopathy-screening-and-counseling-in-the-reproductive-setting-and-fetal-diagnosis
- Benz Jr EJ. Classical thalassemia syndromes (genotypes and laboratory findings). https://www.uptodate.com/contents/image?imageKey=HEME%2F115337&topicKey=HEME%2F7116&search=alpha%20thalassemia&source=outline_link&selectedTitle=1~97
- Steinberg MH. Structure and function of normal hemoglobins. Updated April 2, 2024. https://www.uptodate.com/contents/structure-and-function-of-normal-hemoglobins
- He J, Song W, Yang J, et al. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: the Dai nationality, China. Genetics in medicine : official journal of the American College of Medical Genetics. Sep 2017;19(9):1022-1031. doi:10.1038/gim.2016.218
- Zhang H, Li C, Li J, et al. Next-generation sequencing improves molecular epidemiological characterization of thalassemia in Chenzhou Region, P.R. China. J Clin Lab Anal. 2019;33(4):e22845-e22845. doi:10.1002/jcla.22845
- Shook LM, Haygood D, Quinn CT. Clinical Utility of Confirmatory Genetic Testing to Differentiate Sickle Cell Trait from Sickle-beta(+)-Thalassemia by Newborn Screening. Int J Neonatal Screen. Mar 2020;6(1)doi:10.3390/ijns6010007
- Chen D, Shen X, Xu Y, et al. Successful four-factor preimplantation genetic testing: α- and β-thalassemia, human leukocyte antigen typing, and aneuploidy screening. Systems Biology in Reproductive Medicine. 2021/03/04 2021;67(2):151-159. doi:10.1080/19396368.2020.1832158
- Nosheen A, Ahmad H, Qayum I, Siddiqui N, Abbasi FM, Iqbal MS. Premarital genetic screening for beta thalassemia carrier status of indexed families using HbA2 electrophoresis. JPMA The Journal of the Pakistan Medical Association. Oct 2015;65(10):1047-9.
- Satirapod C, Sukprasert M, Panthan B, et al. Clinical utility of combined preimplantation genetic testing methods in couples at risk of passing on beta thalassemia/hemoglobin E disease: A retrospective review from a single center. PLoS One. 2019;14(11):e0225457. doi:10.1371/journal.pone.0225457
- Fu Y, Shen X, Chen D, Wang Z, Zhou C. Multiple displacement amplification as the first step can increase the diagnostic efficiency of preimplantation genetic testing for monogenic disease for β-thalassemia. J Obstet Gynaecol Res. Aug 2019;45(8):1515-1521. doi:10.1111/jog.14003
- Chen D, Shen X, Wu C, et al. Eleven healthy live births: a result of simultaneous preimplantation genetic testing of α- and β-double thalassemia and aneuploidy screening. J Assist Reprod Genet. 2020;37(3):549-557. doi:10.1007/s10815-020-01732-7
- Dan M, Gutu B-I, Severin E, Tanase V-G. Innovative and Needs-led research on β-thalassemia treatment methods. Frontiers in Hematology. 01/04 2023;1doi:10.3389/frhem.2022.1085952
- Gregg AR, Aarabi M, Klugman S, et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genetics in medicine : official journal of the American College of Medical Genetics. Oct 2021;23(10):1793-1806. doi:10.1038/s41436-021-01203-z
- Langlois S, Ford JC, Chitayat D, et al. Carrier Screening for Thalassemia and Hemoglobinopathies in Canada. Journal of Obstetrics and Gynaecology Canada. 2008;30(10):950-959. doi:10.1016/s1701-2163(16)32975-9
- Désilets V, De Bie I, Audibert F. No. 363-Investigation and Management of Non-immune Fetal Hydrops. Journal of Obstetrics and Gynaecology Canada. 2018;40(8):1077-1090. doi:10.1016/j.jogc.2017.12.011
- Tubman VN, Fung EB, Vogiatzi M, et al. Guidelines for the Standard Monitoring of Patients with Thalassemia: Report of the Thalassemia Longitudinal Cohort. J Pediatr Hematol Oncol. Apr 2015;37(3):e162-9. doi:10.1097/mph.0000000000000307
- ACOG. ACOG Commitee Opinion 691: Carrier Screening for Genetic Conditions. Obstet Gynecol. 2017;129(3):e41-e55. doi:10.1097/AOG.0000000000001952
- ACOG. Hemoglobinopathies in Pregnancy. 2022. https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2022/08/hemoglobinopathies-in-pregnancy
- NHS. SCT Screening: handbook for antenatal laboratories. https://assets.publishing.service.gov.uk/government/uploads/system/ uploads/attachment_data/file/1039978/Referral_guidelines_for_antenatal_screening_specimens_final.pdf
- APHL. Hemoglobinopathies: Current Practices for Screening, Confirmation and Follow-up. Association of Public Health Laboratories; 2015. https://www.cdc.gov/sickle-cell/media/fact-sheets/best-practices/hemoglobinopathies-current-practices-nbs.pdf
- PHE. Sickle cell and thalassaemia screening: commission and provide. Updated January 24, 2022. https://www.gov.uk/government/collections/sickle-cell-and-thalassaemia-screening-commission-and-provide
- Ryan K, Bain BJ, Worthington D, et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. Br J Haematol. Apr 2010;149(1):35-49. doi:10.1111/j.1365-2141.2009.08054.x
- TIF. 2021 Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). 2021;doi:10.1097/HS9.0000000000000732
Coding Section
| Codes |
Number |
Description |
| CPT |
81257 |
HBA1/HBA2 (alpha globin 1 and alpha globin 2) (e.g., alpha thalassemia, Hb Bart hydrops fetalis syndrome, HbH disease), gene analysis, for common deletions or variant (e.g., Southeast Asian, Thai, Filipino, Mediterranean, alpha3.7, alpha4.2, alpha20.5 and Constant Spring) |
|
|
81258 (effective 1/1/2018) |
HBA1/HBA2 (alpha globin 1 and alpha globin 2) (e.g., alpha thalassemia, Hb Bart hydrops fetalis syndrome, HbH disease), gene analysis; known familial variant |
|
|
81259 (effective 1/1/2018) |
HBA1/HBA2 (alpha globin 1 and alpha globin 2) (e.g., alpha thalassemia, Hb Bart hydrops fetalis syndrome, HbH disease), gene analysis; full gene sequence |
|
|
81269 (effective 1/1/2018) |
HBA1/HBA2 (alpha globin 1 and alpha globin 2) (e.g., alpha thalassemia, Hb Bart hydrops fetalis syndrome, HbH disease), gene analysis; duplication/deletion variants |
|
|
81361 |
HBB (hemoglobin, subunit beta) (e.g., sickle cell anemia, beta thalassemia, hemoglobinopathy); common variant(s) (eg, HbS, HbC, HbE) |
|
|
81362 |
HBB (hemoglobin, subunit beta) (e.g., sickle cell anemia, beta thalassemia, hemoglobinopathy); known familial variant(s) |
|
|
81363 |
HBB (hemoglobin, subunit beta) (e.g., sickle cell anemia, beta thalassemia, hemoglobinopathy); duplication/deletion variant(s) |
|
|
81364 |
HBB (hemoglobin, subunit beta) (e.g., sickle cell anemia, beta thalassemia, hemoglobinopathy); full gene sequence |
|
|
S3845 |
Genetic testing for alpha-thalassemia |
|
|
S3846 |
Genetic testing for hemoglobin E beta-thalassemia |
| ICD-9-CM Diagnosis |
V26.31 |
Testing of female genetic disease carrier status |
|
|
V26.34 |
Testing of male for genetic disease carrier status |
| ICD-10-CM (effective 10/01/15) |
Z31.430 |
Encounter of female for testing for genetic disease carrier status for procreative management |
|
|
Z31.440 |
Encounter of male for testing for genetic disease carrier status for procreative management |
| ICD-10-PCS (effective 10/01/15) |
|
Not applicable. ICD-10-PCS codes are only used for inpatient services. There are no ICD procedure codes for laboratory tests. |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other non-affiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 07/17/2025 | Annual review no change to policy intent. Updating description, rationale, and references. Removing CPT 96040 and S0265. |
| 08/01/2024 | Annual review, no change to policy intent. Updating description, table of Terminology, rationale, references. |
| 07/26/2023 | Annual review, no change to policy intent. Policy updated for clarity and consistency. Also updating description including direction to us CAM 358 for prenatal genetic screening issues. Updating table of terminology, rational, and references |
| 07/19/2022 |
Annual review, no change to policy intent. Updating description, rationale and references. |
| 07/20/2021 |
Annual review, no change to policy intent. Updating description, rationale and references. |
| 07/01/2020 |
Annual review, adding policy statement related to genetic counseling. Updating coding. No other changes to policy. |
| 07/12/2019 |
Annual review, updating title and policy to include Beta thalassemia. Also updating coding. |
| 07/18/2018 |
Annual review, no change to policy intent. |
| 12/7/2017 |
Updating policy with 2018 coding. No other changes. |
| 07/20/2017 |
Annual review, rewriting policy verbiage for clarity. No other changes to policy intent. |
| 04/25/2017 |
Interim review to align with Avalon quarterly schedule. Updated category to Laboratory. |
| 11/08/2016 |
Interim review to add medical necessity criteria for testing. No other changes. |
| 04/27/2016 |
Annual review, adding " Genetic testing for patients with hemoglobin H disease (alpha-thalassemia intermedia) to determine prognosis is considered investigation" to policy. No other change to policy intent. Updating background, description, guidelines, rationale and references. |
| 04/15/2015 |
Annual review, no change to policy intent, however, verbiage related to point #2 in policy has been updated for clarity. Preconception (carrier) testing for α-thalassemia in prospective parents may be considered medically |
| 04/09/2014 |
New Policy. |