
Genetic Testing for Alzheimers Disease - CAM 287
Description
Alzheimer disease (AD) is a neurodegenerative disease defined by a gradual decline in memory, cognitive functions, gross atrophy of the brain, and accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles (Karch et al., 2014).
Familial Alzheimer disease (FAD) is a rare, inherited form of AD. FAD has an earlier onset than other forms of Alzheimer disease with symptoms developing in individuals in their thirties or forties. Genetic counseling is strongly recommended for individuals pursuing genetic testing for FAD.
Regulatory Status
On April 6, 2017 the FDA approved the 23andMe PGS Genetic Health Risk Report for Late-onset Alzheimer Disease, indicated for reporting of the ε4 variant in the APOE gene. The report describes if a person's genetic result is associated with an increased risk of developing Late-onset Alzheimer Disease, but it does not describe a person's overall risk of developing Alzheimer Disease. The ε4 variant included in this report is found and has been studied in many ethnicities. Detailed risk estimates have been studied the most in people of European descent (FDA, 2017a).
Other tests for Alzheimer genes are considered laboratory developed tests (LDT); developed, validated and performed by individual laboratories. LDTs are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared these tests; however, FDA clearance or approval is not currently required for clinical use.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- When the results of the testing will inform reproductive decision making, genetic testing for APP, PSEN1, and PSEN2 is considered MEDICALLY NECESSARY for individuals with one of the following conditions:
- For individuals with a family history of autosomal dominant dementia (one or more instances of early-onset Alzheimer disease [AD] [see Note 1]).
- For individuals with a first-degree (see Note 2) relative with a known mutation in the PSEN1, PSEN2, or APP genes.
- For symptomatic individuals (suspected early-onset AD [see Note 1]) with an unknown family history (e.g., adoption).
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- Outside of situations addressed in AHS-M2021-Pharmacogenetic Testing, testing of APOE is considered NOT MEDICALLY NECESSARY.
- For all purposes other than reproductive decision making, genetic testing for AD DOES is considered NOT MEDICALLY NECESSARY, including any of the following situations:
- Testing to confirm a diagnosis of Alzheimer disease (any type).
- Testing for familial Alzheimer disease in individuals less than 18 years of age.
- Testing of any other genes not listed above.
- Testing for purposes of Alzheimer disease risk assessment.
- Screening asymptomatic individuals.
NOTES:
Note 1: Early-onset Alzheimer disease is Alzheimer disease that developed before the individual was 65 years of age.
Note 2: First-degree relatives include parents, full siblings, and children of the individual.
Note 3: For 2 or more gene tests being run on the same platform, please refer to CAM 235 Reimbursement Policy.
Table of Terminology
| Term |
Definition |
| AAN |
American Academy of Neurology |
| AAO |
Age at onset |
| ACMG |
American College of Medical Genetics and Genomics |
| AD |
Alzheimer disease |
| AMP |
Association for Molecular Pathology |
| APOE |
Apolipoprotein E |
| APP |
Amyloid precursor protein |
| CASP7 |
Caspase-7 |
| CLIA’88 |
Clinical Laboratory Improvement Amendments of 1988 |
| CMS |
Centers for Medicare & Medicaid Services |
| CSF |
Cerebrospinal fluid |
| DTC |
Direct to consumer |
| ECRG4 |
Esophageal cancer-related gene 4 |
| EFNS |
European Federation of Neurological Societies |
| ENS |
European Neurological Society |
| EOAD |
Early-onset autosomal dominant Alzheimer disease |
| EOFAD |
Early onset familial Alzheimer disease mutations |
| FAD |
Familial Alzheimer disease |
| FDA |
Food And Drug Administration |
| FTLD |
Frontotemporal lobar degeneration |
| GRN |
Granulin precursor |
| GRS |
Genetic risk score |
| GWAS |
Genome-wide association studies |
| HDAC9 |
Histone deacetylase 9 |
| LDT |
Laboratory-developed test |
| LOAD |
Late-onset Alzheimer disease |
| MAPT |
Microtubule-associated protein tau |
| MPS |
Massive parallel resequencing |
| NGS |
Next-generation sequencing |
| NIA |
National Institute on Aging |
| NSGC |
National Society of Genetic Counselors |
| PGS |
Personal genome service |
| PSEN1 |
Presenilin 1 |
| PSEN2 |
Presenilin 2 |
| USPSTF |
United States Preventive Services Task Force |
Rationale
Alzheimer disease (AD) is a devastating neurodegenerative disease with a strong genetic component and is considered the predominant form of dementia. There are more than 55 million people living with dementia worldwide (WHO, 2023).This number is estimated to increase to 131.5 million by 2050 (Prince, 2016). The average lifetime risk of developing AD is 10% – 12%. This risk at least doubles with the presence of a first-degree relative with the disorder (Goldman et al., 2011). The genetic predisposition of AD, even for late-onset AD patients, is estimated to be 60% – 80% (Gatz et al., 2006).
Most patients develop clinical symptoms after the age of 65 (spontaneous or late-onset AD); however, up to 10% of patients have an earlier onset of disease (early-onset AD) (Kumar, 2018). AD is characterized by severe neuronal loss, aggregation of extracellular amyloid β plaques, and intraneuronal tau protein tangles resulting in progressive deterioration of memory and cognitive functions (Keene, 2023). Enormous burden on public health is due to the high costs associated with care and treatment. Aside from drugs that temporarily relieve symptoms, no treatment currently exists for AD (Van Cauwenberghe et al., 2016).
Autosomal dominant AD is rare (< 1%), but the discovery of fully penetrant pathogenic mutations of Amyloid precursor protein (APP) (Goate et al., 1991; St George-Hyslop et al., 1987), Presenilin 1 (PSEN1) (Sherrington et al., 1995; Van Broeckhoven et al., 1992), and Presenilin 2 (PSEN2) (Sherrington et al., 1996), inherited in an autosomal dominant fashion, has identified molecular mechanisms and pathways involved in AD pathogenesis and valuable targets currently used in diagnosis and drug development (Schneider et al., 2014; Van Cauwenberghe et al., 2016).
One of the primary features of AD is the buildup of amyloid-β protein in the brain. This protein is poisonous to neurons and is normally cleaved by secretases. However, certain genetic mutations may cause these clearing mechanisms to weaken, leading to an overall increase in amyloid-β production. As amyloid-β starts to aggregate in the brain, it creates fibrils that ultimately cause neurological damage such as the characteristic dementia (Keene, 2023).
Amyloid precursor protein (APP) is proteolytically processed in the constitutive pathway by α- and γ-secretases, resulting in nonpathogenic fragments. However, in the amyloidogenic pathway, subsequent proteolysis of APP by β-secretase and γ-secretase gives rise to a mixture of Aβ peptides with different lengths, of which Aβ1–42 are more aggregation-prone and are predominantly present in amyloid plaques in brains of AD patients. A total of 39 APP mutations have been described; all of which affect proteolysis of APP in favor of Aβ1–42 (Cruts et al., 2012).
Presenilin 1 (PSEN1) and Presenilin 2 (PSEN2) are highly homologous genes. Both proteins encoded by these genes are essential components of the γ-secretase complex, which catalyzes the cleavage of membrane proteins, including APP. Mutations in PSEN1 and PSEN2 impair the γ-secretase-mediated cleavage of APP, resulting in an increased proportion of Aβ1–42 (Cruts & Van Broeckhoven, 1998) (Cruts & Van Broeckhoven, 1998). PSEN1 is located on chromosome 14 whereas PSEN2 is located on chromosome 1. However, PSEN1 is generally associated with a worse prognosis; it has full penetrance compared to 95% penetrance for PSEN2, and age of onset was over 10 years earlier for PSEN1 mutations compared to PSEN2 (Ryman et al., 2014; Sherva & Kowall, 2023).
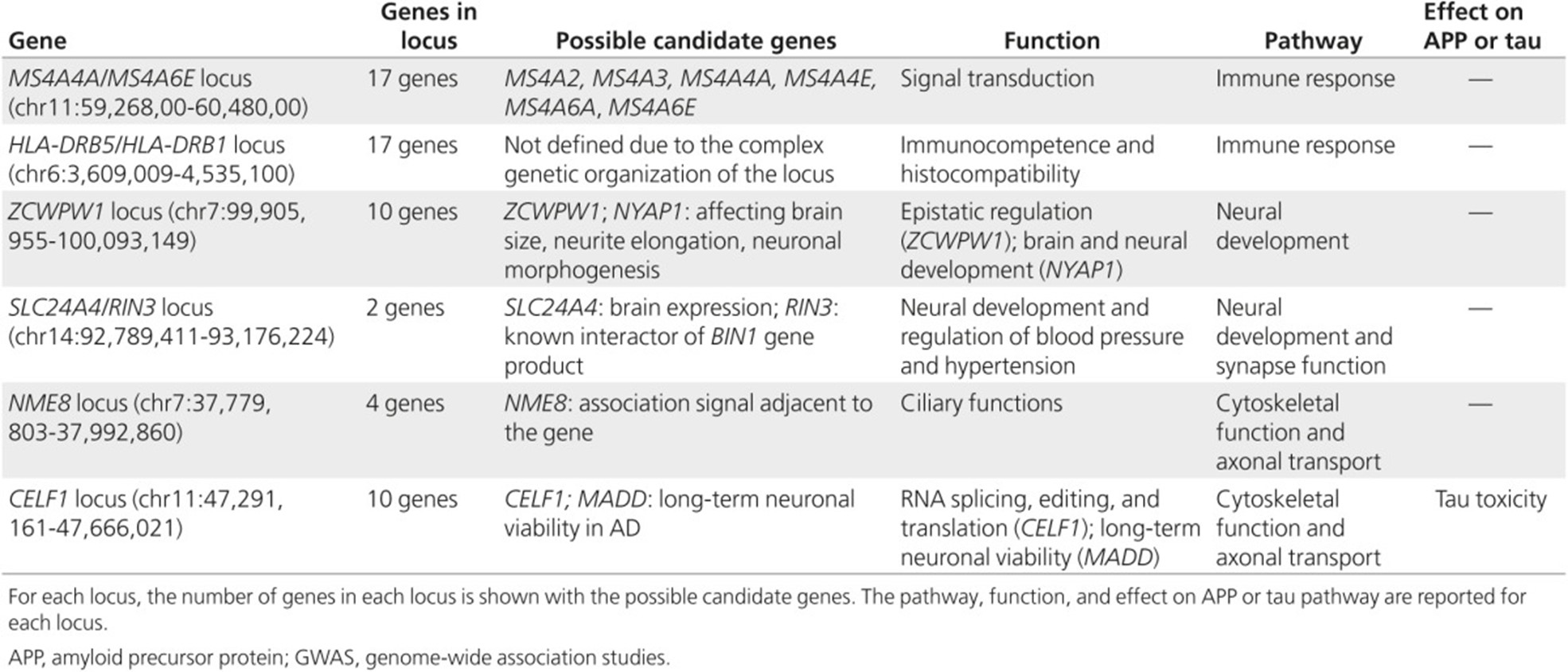
Late-onset AD is considered a multifactorial with a strong but complex genetic predisposition (Gatz et al., 2006) involving gene mutations and polymorphisms that may interact with each other or with environmental factors. The ɛ4 allele of APOE was the only major gene known to increase disease risk for both early-onset and late-onset AD. More recently, genome-wide association studies (GWAS) and massive parallel resequencing (MPS) efforts have identified of at least 21 additional genetic risk loci. These loci, shown in the table below from Van Cauwenberghe et al. (2016), are estimated to explain about 28% of the heritability of liability, 30% of familial risk, and over 50% of sibling recurrence risk of developing AD (Van Cauwenberghe et al., 2016). Researchers have recently identified a rare missense variant in the CASP7 gene that may be associated with familial late-onset AD (Zhang et al., 2019), as well as a T allele of the CD33 rs3865444 polymorphism also associated with late-onset AD (Mehdizadeh et al., 2019).
The APOE gene has several alleles, with the ɛ4 allele contributing to an increased risk of late-onset AD and the ɛ2 allele contributing to a decreased risk of late-onset AD compared to the common APOE ɛ3 allele (Yamazaki et al., 2019). Researchers now report that APOE influences tau pathology as well as neurodegeneration mediated by tau and microglial responses to AD pathologies; further, APOE ɛ4 is “either pathogenic or shows reduced efficiency in multiple brain homeostatic pathways, including lipid transport, synaptic integrity and plasticity, glucose metabolism and cerebrovascular function” (Yamazaki et al., 2019).
In 2023, the FDA approved Lecanemab (brand name Leqembi), an amyloid beta-directed antibody, for the treatment of Alzheimer Disease in adult patients (FDA, 2023). One potential side effect of Leqembi is amyloid related imaging abnormalities (ARIA), which may be more likely to occur in people who are homozygous APOE ε4 carriers (Leqembi, 2024). The FDA cautions that “the prescribing information states that testing for ApoE ε4 status should be performed before starting treatment with Leqembi to inform the risk of developing ARIA” (FDA, 2023).
Proprietary Testing
Early-Onset Alzheimer Disease
Several companies have developed hereditary AD panels. The Invitae Hereditary Alzheimer Disease Panel tests for three genes associated with early-onset hereditary AD: APP, PSEN1 and PSEN2. This test may utilize a blood, DNA or saliva sample and has a 10 – 21 day turnaround time (Invitae, 2023). The ADmark® Early Onset Alzheimer's Evaluation also tests for the three known early-onset hereditary AD genes: APP, PSEN1 and PSEN2. This test detects sequence variants in these genes, as well as duplications in the APP gene. A whole blood sample is required, and a turnaround time of 21-28 days can be expected (Athena, 2023b).
Another panel by Fulgent, termed the Parkinson-Alzheimer-Dementia NGS panel, tests for 35 genes that are associated with developing Parkinson disease, Alzheimer disease and dementia. Some of the genes tested in this panel include APOE, APP, PSEN1 and PSEN2. This test also requires a blood sample or buccal swab and has a three to five week turnaround time (Fulgent, 2023).
Roche has developed the Elecsys® Amyloid Plasma Panel, which has received FDA Breakthrough Device Designation. According to Roche, it is the first qualitative test that combines the result of phosphorylated Tau (pTau) 181 protein assay and APOE ɛ4 in human plasma. This can leader to earlier diagnosis of Alzheimer’s disease as patients “testing negative with the Elecsys Amyloid Plasma Panel are unlikely to be amyloid positive and should be investigated for other causes of cognitive decline” (Roche, 2024).
Late-Onset Alzheimer Disease
Athena diagnostics developed the ADmark® ApoE Genotype Analysis and Interpretation test which detects APOE ɛ2, ɛ3, ɛ4 alleles using restriction fragment length polymorphism (Athena, 2023a). Athena will not perform this test on individuals younger than 18 years of age and recommends pre and post-test genetic counseling; a whole blood sample is required, and a turnaround time of 7 – 14 days can be expected.
Clinical Utility and Validity
Early-Onset Alzheimer Disease
Comprehensive genetic counseling protocols are available for AD diagnostic and predictive testing to provide a framework for clinicians and geneticists to evaluate which patients may benefit from genetic testing. Available genetic diagnostic and predictive screening for causal mutations of early-onset AD in APP, PSEN1, and PSEN2 are only responsible for a small portion of AD patients’ risk. They account for approximately 60% – 70% of familial autosomal dominant AD, but less than 10 percent of early-onset AD and less than one percent of AD overall (Sherva & Kowall, 2023). For a significant number of patients for whom genetic diagnostic screening is requested, the tests will be negative without excluding a genetic cause of disease (Van Cauwenberghe et al., 2016). Furthermore, the identification of a mutation is not a certain predictor of disease or onset age, given that these mutations can vary in terms of penetrance and gene expression. Nevertheless, the ability to identify an explanation for the clustering of AD in a family and the ability to use this toward predictive testing in subsequent generations provide an important step toward autonomy of patients and at-risk individuals (Van Cauwenberghe et al., 2016). Testing for these highly penetrant mutations often carries significant personal and familial utility which the ACMG (American College of Medical Genetics) has recently supported as important clinical utilities (ACMG, 2015). New mutations in the APP, PSEN1, and PSEN2 genes are constantly being identified. For example, two probable pathogenic variants, PSEN2 p.A415S and p.M174I, were identified by Wong et al. (2020).
Janssen et al. (2003) aimed to determine the proportion of patients with early-onset AD with a positive family history that had mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes. A mutational analysis was performed in 31 probands with probable or definite AD from UK families (age at onset < 61 years). A total of 23 patients fulfilled criteria for autosomal dominant inheritance. In 17 (55%) probands the authors identified eight novel PSEN1 sequence variants and eight recognized pathogenic mutations. In four (13%) probands the authors identified one novel APP sequence variant (H677R) and two recognized mutations. Further, 21 of 31 (68%) probands were associated with a sequence variant in APP or PSEN1. Nine of the 11 (82%) probands with neuropathologically confirmed AD who additionally fulfilled recognized criteria for autosomal dominant inheritance were associated with a sequence variant in APP or PSEN1. The 10 patients in whom the authors were unable to identify a mutation in APP, PSEN1, or PSEN2 were older than the probands with sequence variants (55.4 vs 44.7 years, respectively). The authors concluded that sequence variants in APP and PSEN1 accounted for the majority of neuropathologically confirmed autosomal dominant early-onset AD (Janssen et al., 2003).
Shea et al. (2016) conducted a study to assess the differences in clinical presentations of different genotypes of FAD. A total of 658 pedigrees were evaluated. The authors found that patients with PSEN1 mutations tended to have earlier age of onset than either PSEN2 or APP mutations. Patients with PSEN1 were also more commonly affected by symptoms such as seizures or myoclonus, whereas patients with PSEN2 mutations were more commonly affected by disorientation. Patients with APP mutations were more likely to present with aggression or apraxia (Shea et al., 2016).
Lanoiselee et al. (2017) completed a large genetic screening study of familial and sporadic cases of APP, PSEN1, and PSEN2 mutations in early-onset AD. Data was taken from 23 French hospitals from 1993 onward; the total number of families identified with mutations was 170 (these families were required to have two first-degree relatives with early-onset AD with an age of onset ≤65 years). One hundred and twenty-nine sporadic cases were also screened with an age of onset ≤ 51 years. The authors not that “APP, PSEN1, or PSEN2 mutations were identified in 53 novel AD-EOAD [early-onset AD] families. Of the 129 sporadic cases screened, 17 carried a PSEN1 mutation and one carried an APP duplication (13%); this led to the conclusion that a portion of PSEN1 mutations occur de novo (Lanoiselee et al., 2017).
Giau et al. (2019) screened 67 de novo early-onset AD cases by next-generation sequencing (NGS) to identify pathogenic variants linked to neurodegenerative disorders in the Korean population. They were able to find three missense mutations in PSEN1 and a variant in PSEN2 within 6% of the cases with early onset AD, but also found “67 missense mutations in susceptibility genes for late-onset AD … which may be involved in cholesterol transport, inflammatory response, and β-amyloid modulation.” They also found “70 additional novel and missense variants in other genes, such as MAPT, GRN, CSF1R, and PRNP, related to neurodegenerative diseases, which may represent overlapping clinical and neuropathological features with AD.” Multiple rare variants were found among this patient population as well (Giau et al., 2019).
Qin et al. (2020) conducted an analysis on the genotype and phenotype correlation for early onset familial AD in a Chinese population. With respect to specific mutations, the researchers found that for APP mutations, the clinical phenotype was relatively heterogeneous, with an average age at onset ranging from the 40s-50s and clinical presentations of “cognitive dysfunction, especially executive dysfunction and disorientation. Extrapyramidal signs, behavioral, and psychiatric symptoms could also be detected in Chinese APP EOFAD [early onset familial Alzheimer disease mutations].” For those with PSEN1 mutations, the age at onset was in the early 40s, and with an amnestic cognitive profile, as well as myoclonus and seizures. “Extrapyramidal signs, behavioral, and psychiatric symptoms (anxiety, hallucinations, delusions) and ataxia are significantly more frequently found in EOFAD with PSEN1 mutations.” PSEN2 mutations were found to be the least common, and “compared to PSEN1 mutation carriers, carriers with PSEN2 mutations have a later AAO, relatively longer disease duration and a more variable disease expression.” Overall, the researchers found that most of the mutations in China were novel in comparison to pathogenic variants among Caucasians (Qin et al., 2020).
Late-Onset Alzheimer Disease
The primary gene associated with late-onset AD is the apolipoprotein E (APOE) gene on chromosome 19, particularly its epsilon (ɛ) allele. This apolipoprotein is thought to play a role in cholesterol homeostasis and aid in removal of the amyloid- β protein that is at the core of AD. There are three isoforms of this allele: ɛ2, ɛ3, and ɛ4. The ɛ4 allele binds much more rapidly to the amyloid protein; however, it is less efficient than the other two alleles in protein transfer. These characteristics combined have made the ɛ4 allele a potential genetic risk factor of AD (Sherva & Kowall, 2023).
The role of genetics in diagnosis and risk prediction in late-onset AD is much less straightforward. APOE ɛ4 is associated with changes in lipid metabolomics in AD patients, and is likely a factor in even the early stages of AD development (Pena-Bautista et al., 2020). Further, it has been suggested that APOE ɛ4 is a selective risk factor, affecting memory-related AD manifestations of the disease more than language-related implications (Weintraub et al., 2020). Despite the established evidence of APOE ɛ4 as a risk factor for AD, its value in disease prediction in a clinical setting is limited, and the relevance of clinical testing for common genetic variations identified in GWAS is even more limited. Combining multiple susceptibility loci into a global genetic risk score (GRS) might improve the prediction of individuals at risk. However, the most comprehensive risk prediction model developed to date only achieved a sensitivity of 55% and a specificity of 78%, impeding use in clinical practice (de Calignon et al., 2012; Van Cauwenberghe et al., 2016).
Naj et al. (2014) assessed the effect of APOE alleles on average age of onset in AD patients. Fourteen studies containing 9,162 patients were examined, and the APOE allele was found to contribute 3.9% of the variation of age of onset. Each copy of the ε4 was found to reduce the age of onset by 2.45 years (Naj et al., 2014).
Cohn-Hokke et al. (2017) examined the social and personal effects of testing for hereditary neurodegenerative diseases from 74 patient survey responds. The authors concluded that “the result of predictive testing on adult-onset neurodegenerative diseases does not have a large negative effect on social and personal life, although these observations should be interpreted with caution because of the small number of participants and low response rate” (Cohn-Hokke et al., 2017).
The ancestral APOE ɛ4 risk of AD has been studied across Puerto Rican and African American populations. A total of 1,986 participants with late-onset AD (1,766 African Americans and 220 Puerto Ricans) and 3,899 healthy controls older than 65 years of age (3,730 African Americans and 169 Puerto Ricans) participated in this study. The authors note that “APOE ε4 alleles on an African background conferred a lower risk than those with a European ancestral background, regardless of population” (Rajabli et al., 2018). This study shows that the risk conferred by the APOE ε4 allele differs across populations; the cause of this risk is unknown but may be due to genetic variation, environmental factors, or cultural factors associated with ancestry. Stevenson-Hoare et al. (2023) studied the plasma biomarkers and genetics used for AD diagnosis. The study included 1439 people with AD, with a mean age of 68 years, and 508 controls, with a mean age of 82 years. The authors measured plasma concentrations of 40 and 42 amino acid-long amyloid β fragments, tau phosphorylated at amino acid 181, neurofilament light, and glial fibrillary acidic protein. “The prediction accuracy of Alzheimer’s disease clinical diagnosis by the combination of all biomarkers, APOE and polygenic risk score reached area under receiver operating characteristic curve (AUC) = 0.81” The genetic risk was compared to age of onset and disease duration. “All biomarkers were significantly associated with age in cases and controls.” The authors conclude that “biomarker-based diagnosis is not perfect because the biomarker measurements in older controls are similar to those in younger clinically diagnosed AD cases” but, “blood plasma biomarkers can only be a useful tool for the assessment and prediction of AD in the context of other genetic and/or clinical information” (Stevenson-Hoare et al., 2023).
There is an ethical concern for mental and personal wellbeing after testing for late-onset AD. Pavarini et al. (2021) interviewed 31 people aged 16 to 26 whose grandparents had late-onset AD. The interviews focused on the participant’s moral attitudes and motivations towards online and direct-to-consumer genetic testing for late-onset AD. The authors found that participants agreed that “people should have the right to access these services,” but were concerned about “potential distress in response to learning about risk.” Overall, face-to-face services were preferred over online. The authors suggest that these results highlight the importance of support, particularly to young people, before and after testing (Pavarini et al., 2021).
Pavarini et al. (2021) studied the psychological impact of genetic testing on healthy individuals at risk for AD. This study included individuals that underwent counseling and testing protocol. The authors performed a psychological assessment at zero, six and 12 months following genetic test results on 24 participants from 13 families. The authors found that carriers of pathogenic variants (PSEN1, PSEN2, GRN) showed higher scores than noncarriers on the Resilience Scale for Adults, social competence, and the Multidimensional Health Locus of Control. The authors conclude that “at-risk individuals undergoing predictive testing showed benefit on personal life and no detrimental impact on a broad range of psychological outcomes” (Galluzzi et al., 2022).
Largent et al. (2021) studied the ethical, social, and legal concerns regarding sharing AD genetic testing results. The authors predict that genetic testing for AD in people who are not yet cognitively impaired will become more common in clinical practice. The authors analyzed the decisions to share genetic information of participants in the A4 Study and the API Generation Program, two clinical trials that shared genetic results with the participants. Participants with no elevated amyloid or no APOE ε4 alleles did not pay close attention to the decision regarding sharing this information, and they felt it was good or no news. Participants with elevated amyloid or APOE ε4 alleles felt the information was sensitive and burdensome to decide if they should share or hide it from others. These individuals felt more worried about stigmatization and discrimination, even though they were not currently cognitively impaired. Additionally, close friends and family members of these individuals are more likely to take up a “pre-caregiver” role, potentially changing their relationship; “12% of participants at increased risk for dementia stated that they disclosed their result so that they could ‘count on other people to tell me if I’m changing.’” The authors state that these results and concerns “highlight the need for additional legal protections and policy changes in anticipation of the coming transformation of AD clinical care” (Largent et al., 2021).
American College of Medical Genetics and Genomics (ACMG) and National Society of Genetic Counselors (NSGC)
The American College of Medical Genetics and Genomics (ACMG) and the National Society of Genetic Counselors (NSGC) issued joint practice guidelines related to the genetic assessment of AD. These guidelines include the following recommendations (Goldman et al., 2011):
- “Pediatric testing for AD should not occur.”
- “Prenatal testing for AD is not advised if the patient intends to continue a pregnancy with a mutation.”
- “Genetic testing for AD should only occur in the context of genetic counseling (in-person or through videoconference) and support by someone with expertise in this area. Symptomatic patients: Genetic counseling for symptomatic patients should be performed in the presence of the individual’s legal guardian or family member.”
- “DTC (direct to consumer) APOE testing is not advised.”
- “A risk assessment should be performed by pedigree analysis to determine whether the family history is consistent with EOAD [early-onset AD] or LOAD (late-onset AD) and with autosomal dominant (with or without complete penetrance), familial or sporadic inheritance.”
For families in which an autosomal dominant AD gene mutation is a possibility:
- “Testing for genes associated with early-onset autosomal dominant AD should be offered in the following situations:
- “A symptomatic individual with EOAD in the setting of a family history of dementia or in the setting of an unknown family history (e.g., adoption).
- “Autosomal dominant family history of dementia with one or more cases of EOAD.”
- “A relative with a mutation consistent with EOAD (currently PSEN1/2 or APP).”
- “Ideally, an affected family member should be tested first. If no affected family member is available for testing and an asymptomatic individual remains interested in testing despite counseling about the low likelihood of an informative result (a positive result for a pathogenic mutation), he/she should be counseled according to the recommended protocol. If the affected relative, or their next of kin, is uninterested in pursuing tested, the option of DNA banking should be discussed.”
For families in which an autosomal dominant AD is unlikely:
- “Discuss that both sporadic and familial cases can be due to a genetic susceptibility. Risk estimates are only available for first-degree relatives of an affected individual in sporadic or familial cases.”
- “Genetic testing for susceptibility loci (e.g., APOE) is not clinically recommended due to limited clinical utility and poor predictive value. If a patient wishes to pursue testing despite genetic counseling and recommendations to the contrary, testing may be considered at the clinician’s discretion.”
Finally, the authors comment that “in general, clear genotype-phenotype correlations cannot typically be made for the three causative genes, and age of onset can vary more than 20 years within the same family” (Goldman et al., 2011).
In 2019, an addendum was published for the 2011 guidelines. The ACMB board of directors reaffirmed these guidelines (as of June 25, 2018) with two changes:
- “To use the phrase “pathogenic variant” rather than the word “mutation” in discussing pathogenic variants related to autosomal dominant early-onset Alzheimer disease. This would be consistent with current ACMG/AMP Guidelines for Variant Interpretation and Reporting.
- Because this document no longer meets the criteria for an evidence-based practice guideline by either the American College of Medical Genetics and Genomics (ACMG) or National Society of Genetic Counselors (NSGC), NSGC reclassified this document as a Practice Resource in 2016, and ACMG is also classifying it as a Practice Resource as of this reaffirmation” (Goldman et al., 2019).
American College of Medical Genetics and Genomics (ACMG)
In the Choosing Wisely Initiative, the ACMG recommended “Don’t order APOE genetic testing as a predictive test for Alzheimer's disease.” The rationale for the recommendation is that “APOE is a susceptibility gene for later-onset Alzheimer disease (AD), the most common cause of dementia. The presence of an ε4 allele is neither necessary nor sufficient to cause AD. The relative risk conferred by the ε4 allele is confounded by the presence of other risk alleles, gender, environment and possibly ethnicity. APOE genotyping for AD risk prediction has limited clinical utility and poor predictive value” (ACMG, 2016).
American Academy of Neurology (AAN)
In 2001 (reaffirmed in 2004), AAN made the following recommendation on the use of genetic testing for Alzheimer’s disease:
- Routine use of APOE genotyping in patients with suspected AD is not recommended at this time (Guideline).
- There are no other genetic markers recommended for routine use in the diagnosis of AD (Guideline) (Knopman et al., 2001).
National Institute on Aging (NIA)
In 2011, Alzheimer’s Disease diagnostic guidelines were revised including latest research results and better scientific understanding of the disease. The development of the new guidelines was led by the National Institute of Health and the Alzheimer’s Association. Diagnostic criteria for Alzheimer’s disease were re-defined. In respect to genetic testing, NIA issued following guidance and recommendations: “A rare type of familial Alzheimer’s disease, called Early-Onset Alzheimer’s Disease (EOAD), is caused by mutations in the amyloid precursor protein, presenilin 1, or presenilin 2 genes. A person who inherits any of these mutations from a parent will almost surely develop Alzheimer’s dementia before age 65. Genetic testing for the disease is common in families with a history of EOAD”; “The major genetic risk factor for the more common, sporadic form of the disease, or Late-Onset Alzheimer’s disease (LOAD), is the ε4 allele of the APOE gene. But carrying this allele by itself does not mean a person has or will develop Alzheimer’s dementia, so genetic testing for APOE ε4 is not recommended outside of a research setting” (NIH, 2022).
The NIA and Alzheimer’s Association released a joint research framework in 2018. In that framework, they state that “Genetics is not formally included in the research framework because our concept of disease rests on neuropathologic change (that can be detected by biomarkers). In contrast, gene variants do not measure pathologic change but rather indicate an individual's risk for developing pathologic change” (Jack et al., 2018).
European Federation of Neurological Societies (EFNS) and European Neurological Society (ENS)
The EFNS and ENS have developed guidelines for the diagnosis and management of disorders associated with dementia. Regarding genetic testing, these guidelines state that “No studies have addressed the value of genetic counselling for patients with dementia or their families when autosomal‐dominant disease is suspected. Because the genetics of dementing illnesses is a very young field, expertise in genetic counselling for the dementias of the elderly is likely to be found only in specialized dementia research centres (Good Practice Point). Screening for known pathogenic mutations can be undertaken in patients with appropriate phenotype or a family history of an autosomal‐dominant dementia. This should only be undertaken in specialist centres with appropriate counselling of the patient and family caregivers, and with consent (Good Practice Point). Pre‐symptomatic testing may be performed in adults where there is a clear family history, and when there is a known mutation in an affected individual to ensure that a negative result is clinically significant. (Good Practice Point)” (Sorbi et al., 2012).
European Federation of Neurological Sciences (EFNS)
In 2010, EFNS published revised recommendations on the diagnosis and management of Alzheimer disease. It stated that “the ApoE 4 allele is the only genetic factor consistently implicated in late-onset AD, but it is neither necessary nor sufficient for development of the disease. Hence, there is no evidence to suggest ApoE testing is useful in a diagnostic setting.” The EFNS recommended that “screening for known pathogenic mutations can be undertaken in patients with appropriate phenotype or a family history of an autosomal dominant dementia. Routine Apo E genotyping is not recommended” (Hort et al., 2010).
United States Preventive Services Task Force (USPSTF)
The USPSTF has concluded that “the current evidence is insufficient to assess the balance of benefits and harms of screening for cognitive impairment in older adults” (Owens et al., 2020).
Alzheimer’s Disease and Related Disorders Therapeutics Work Group
In 2023, a workgroup developed and published information on Lecanemab (Leqembi®) as an approved treatment for Alzheimer disease (AD). The drug can be initiated in early Alzheimer disease for those with confirmed brain amyloid pathology. The working group recommends that patients requiring anticoagulants should not receive lecanemab until more data regarding the interaction with anticoagulants is available. This is because adverse events of amyloid related imaging abnormalities (ARIA) can result with lecanemab and these may involve microhemorrhages and rare macrohemorrhages; in other words, anticoagulation may increase the risk of hemorrhage.
Other recommendations:
- “We recommend that all patients being considered for lecanemab therapy meet diagnostic criteria for mild cognitive impairment (MCI) due to AD or probable mild AD dementia with biomarker evidence (amyloid PET or CSF) of the AD pathophysiological process.”
- “Individuals with depression who scored >8 on the Geriatric Depression Scale or had a body mass index (BMI) greater than 35 or less than 17 were excluded from both the phase 2 and 3 studies. We recommend adopting these same criteria while allowing medical judgment for individual circumstances when the impact of these modifications has been considered.”
- “Patients who are apolipoprotein E ε4 (APOE4) gene carriers, especially APOE4 homozygotes, are at higher risk for ARIA, and the AUR recommends APOE genotyping to better inform risk discussions with patients who are lecanemab candidates.”
- “We recommend excluding patients with any history of seizures until additional data are available.”
- “Cerebral amyloid angiopathy-related inflammation/amyloid beta-related angiitis (CAA-ri/ABRA) increase the risk for ARIA (discussed below) and should exclude patients as treatment candidates.”
- “Patients should not be given lecanemab if they are receiving aducanumab or had severe or recurrent ARIA with use of aducanumab.”
- “Participants with clotting disorders should be excluded.”
“Evidence of cerebral contusion, encephalomalacia, brain aneurysms or other vascular malformations, central nervous system (CNS) infection, and brain tumors other than meningioma or arachnoid cysts excluded patients from phase 3 trial participation. These same restrictions should apply when considering patients for treatment with lecanemab” (Cummings et al., 2023).
International Working Group (IWG)
The IWG released recommendations for the clinical diagnosis of AD in their “Personal View” article responding to the National Institute of Aging and the Alzheimer Association biologically defining AD based on biomarkers. The IWG contests this definition, stating that some cognitively unimpaired people with AD biomarkers may never develop clinical symptoms of AD, and that AD can develop as a co-morbidity of other brain diseases. The IWG goes on to recommend how biomarkers should and should not be used to clinically diagnose AD. “We recommend that AD diagnosis be restricted to the occurrence of positive biomarkers together with specific AD phenotypes, whereas biomarker positive cognitively unimpaired individuals should be considered only at-risk for progression to AD” (Dubois et al., 2021). The IWG recommends:
-
- A “clinical-biological” diagnosis of AD requiring the presence of both a clinical phenotype and a biomarker.
- The common clinical phenotypes for AD are “the amnestic syndrome of the hippocampal type (typical), the posterior cortical atrophy variant, and the logopenic variant primary progressive aphasia.” Less common phenotypes include “behavioural/dysexecutive variant7, the cortico-basal variant, and the other variants of primary progressive aphasia.”
- Both positive amyloid and tau biomarkers must both be present, with the common phenotypes, for AD diagnosis. Uncommon phenotypes with positive AD markers do not ensure an AD diagnosis, a clinician must determine that AD is the dominant pathology.
- “Recommended biomarker measures for amyloid pathology are low CSF Aβ42, increased CSF Aβ40/42 ratio, or increased tracer retention in amyloid PET; for tau pathology, we recommend high CSF p-tau (not total tau [t-tau] due to its lack of specificity) or increased ligand retention in tau PET.”
- The diagnosing clinician must be an expert in critically assessing both clinical and biomarker results.
- “CSF investigation is prioritized as it provides simultaneous information on the 2 types of biomarkers (amyloid and tau) and is less expensive than amyloid-PET and/or tau-PET. Where lumbar puncture is contraindicated, PET investigations are an alternative consideration.”
- Clinical pathological biomarker investigation is not recommended for cognitively unimpaired individuals. This may change in the future with prevention programs.
- When biomarker investigation has been performed on cognitively unimpaired individuals, the IWG proposed a risk stratification system to distinguish an absolute risk group, a high-risk group, and an undefined risk group. Individuals should be counselled before AD biomarker investigation and if they are at-risk for progression to prodromal AD or AD dementia.
- Isolated subjective memory complaints or cognitive decline that are not supported by cognitive impairment are not enough to be part an AD phenotype.
- When AD is co-morbid with other brain pathologies, it is recommended that physicians rely more on the phenotype and follow-up for the diagnosis.
- Physicians should objectively evaluate the added-value of biomarker investigation in symptomatic patients based on the clinical situation, life context, patient wishes, the possibility of participating in disease-modifying trials, and the current global appreciation of how this information can change patient management.
- When pathophysiological biomarkers are unavailable, patients should have a clinical syndrome diagnosis and more attention should be given to rule out non-degenerative causes (Dubois et al., 2021).
References:
- ACMG. (2015). Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med, 17(6), 505-507. https://doi.org/10.1038/gim.2015.41
- ACMG. (2016, September 15). Don’t order APOE genetic testing as a predictive test for Alzheimer disease. https://www.aafp.org/pubs/afp/collections/choosing-wisely/282.html
- Athena. (2023a). ADmark® ApoE Genotype Analysis and Interpretation (Symptomatic). https://www.athenadiagnostics.com/view-full-catalog/admark-apoe-genotype-analysis-and-interpretation-symptomatic1
- Athena. (2023b). ADmark® Early Onset Alzheimer's Evaluation. https://www.athenadiagnostics.com/view-full-catalog/admark-alzheimers-evaluation1
- Cohn-Hokke, P. E., van Swieten, J. C., Pijnenburg, Y. A. L., Tibben, A., Meijers-Heijboer, H., & Kievit, A. (2017). The Effect of Predictive Testing in Adult-Onset Neurodegenerative Diseases on Social and Personal Life. J Genet Couns. https://doi.org/10.1007/s10897-017-0195-3
- Cruts, M., Theuns, J., & Van Broeckhoven, C. (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat, 33(9), 1340-1344. https://doi.org/10.1002/humu.22117
- Cruts, M., & Van Broeckhoven, C. (1998). Presenilin mutations in Alzheimer's disease. Hum Mutat, 11(3), 183-190. https://pubmed.ncbi.nlm.nih.gov/9521418/
- Cummings, J., Apostolova, L., Rabinovici, G. D., Atri, A., Aisen, P., Greenberg, S., Hendrix, S., Selkoe, D., Weiner, M., Petersen, R. C., & Salloway, S. (2023). Lecanemab: Appropriate Use Recommendations. J Prev Alzheimers Dis, 10(3), 362-377. https://doi.org/10.14283/jpad.2023.30
- de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., Pitstick, R., Sahara, N., Ashe, K. H., Carlson, G. A., Spires-Jones, T. L., & Hyman, B. T. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron, 73(4), 685-697. https://doi.org/10.1016/j.neuron.2011.11.033
- Dubois, B., Villain, N., Frisoni, G. B., Rabinovici, G. D., Sabbagh, M., Cappa, S., Bejanin, A., Bombois, S., Epelbaum, S., Teichmann, M., Habert, M. O., Nordberg, A., Blennow, K., Galasko, D., Stern, Y., Rowe, C. C., Salloway, S., Schneider, L. S., Cummings, J. L., & Feldman, H. H. (2021). Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. Lancet Neurol, 20(6), 484-496. https://doi.org/10.1016/s1474-4422(21)00066-1
- FDA. (2017a). DECISION SUMMARY. https://www.accessdata.fda.gov/cdrh_docs/reviews/den160026.pdf
- FDA. (2017b). Decision Summary for 23andMe PGS Genetic Health Risk Report. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/DEN160026.pdf
- Fulgent. (2023). Parkinson-Alzheimer-Dementia NGS Panel. https://www.fulgentgenetics.com/Parkinson-Alzheimer-Dementia
- Galluzzi, S., Mega, A., Di Fede, G., Muscio, C., Fascendini, S., Benussi, L., Tagliavini, F., Frisoni, G. B., & Di Maria, E. (2022). Psychological Impact of Predictive Genetic Testing for Inherited Alzheimer Disease and Frontotemporal Dementia: The IT-DIAfN Protocol. Alzheimer Dis Assoc Disord. https://doi.org/10.1097/wad.0000000000000494
- Gatz, M., Reynolds, C. A., Fratiglioni, L., Johansson, B., Mortimer, J. A., Berg, S., Fiske, A., & Pedersen, N. L. (2006). Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry, 63(2), 168-174. https://doi.org/10.1001/archpsyc.63.2.168
- Giau, V. V., Bagyinszky, E., Yang, Y. S., Youn, Y. C., An, S. S. A., & Kim, S. Y. (2019). Genetic analyses of early-onset Alzheimer's disease using next generation sequencing. Scientific reports, 9(1), 8368-8368. https://doi.org/10.1038/s41598-019-44848-2
- Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., James, L., & et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature, 349(6311), 704-706. https://doi.org/10.1038/349704a0
- Goldman, J. S., Hahn, S. E., Catania, J. W., LaRusse-Eckert, S., Butson, M. B., Rumbaugh, M., Strecker, M. N., Roberts, J. S., Burke, W., Mayeux, R., & Bird, T. (2011). Genetic counseling and testing for Alzheimer disease: Joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med, 13(6), 597-605. https://doi.org/10.1097/GIM.0b013e31821d69b8
- Goldman, J. S., Hahn, S. E., Catania, J. W., LaRusse-Eckert, S., Butson, M. B., Rumbaugh, M., Strecker, M. N., Roberts, J. S., Burke, W., Mayeux, R., & Bird, T. (2019). ADDENDUM: Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med, 21(10), 2404. https://doi.org/10.1038/s41436-019-0559-1
- Hort, J., O'Brien, J. T., Gainotti, G., Pirttila, T., Popescu, B. O., Rektorova, I., Sorbi, S., & Scheltens, P. (2010). EFNS guidelines for the diagnosis and management of Alzheimer's disease. Eur J Neurol, 17(10), 1236-1248. https://doi.org/10.1111/j.1468-1331.2010.03040.x
- Invitae. (2023). Invitae Hereditary Alzheimer's Disease Panel. https://www.invitae.com/en/physician/tests/03504/
- Jack, C. R., Jr., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., Holtzman, D. M., Jagust, W., Jessen, F., Karlawish, J., Liu, E., Molinuevo, J. L., Montine, T., Phelps, C., Rankin, K. P., Rowe, C. C., Scheltens, P., Siemers, E., Snyder, H. M., . . . Silverberg, N. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 14(4), 535-562. https://doi.org/10.1016/j.jalz.2018.02.018
- Janssen, J. C., Beck, J. A., Campbell, T. A., Dickinson, A., Fox, N. C., Harvey, R. J., Houlden, H., Rossor, M. N., & Collinge, J. (2003). Early onset familial Alzheimer's disease: Mutation frequency in 31 families. Neurology, 60(2), 235-239. http://n.neurology.org/content/60/2/235.long
- Karch, C. M., Cruchaga, C., & Goate, A. M. (2014). Alzheimer's disease genetics: from the bench to the clinic. Neuron, 83(1), 11-26. https://doi.org/10.1016/j.neuron.2014.05.041
- Keene, C. D., Montine, Thomas, Kuller, Lewis. (2023, January 19). Epidemiology, pathology, and pathogenesis of Alzheimer disease. https://www.uptodate.com/contents/epidemiology-pathology-and-pathogenesis-of-alzheimer-disease
- Knopman, D. S., DeKosky, S. T., Cummings, J. L., Chui, H., Corey-Bloom, J., Relkin, N., Small, G. W., Miller, B., & Stevens, J. C. (2001). Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology, 56(9), 1143-1153. https://doi.org/10.1212/wnl.56.9.1143
- Kumar, A., Tsao, Jack. (2018). Alzheimer Disease. https://www.ncbi.nlm.nih.gov/books/NBK499922/
- Lanoiselee, H. M., Nicolas, G., Wallon, D., Rovelet-Lecrux, A., Lacour, M., Rousseau, S., Richard, A. C., Pasquier, F., Rollin-Sillaire, A., Martinaud, O., Quillard-Muraine, M., de la Sayette, V., Boutoleau-Bretonniere, C., Etcharry-Bouyx, F., Chauvire, V., Sarazin, M., le Ber, I., Epelbaum, S., Jonveaux, T., . . . Campion, D. (2017). APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med, 14(3), e1002270. https://doi.org/10.1371/journal.pmed.1002270
- Largent, E. A., Stites, S. D., Harkins, K., & Karlawish, J. (2021). 'That would be dreadful': The ethical, legal, and social challenges of sharing your Alzheimer's disease biomarker and genetic testing results with others. J Law Biosci, 8(1), lsab004. https://doi.org/10.1093/jlb/lsab004
- Mehdizadeh, E., Khalaj-Kondori, M., Shaghaghi-Tarakdari, Z., Sadigh-Eteghad, S., Talebi, M., & Andalib, S. (2019). Association of MS4A6A, CD33, and TREM2 gene polymorphisms with the late-onset Alzheimer's disease. Bioimpacts, 9(4), 219-225. https://doi.org/10.15171/bi.2019.27
- Naj, A. C., Jun, G., Reitz, C., Kunkle, B. W., Perry, W., Park, Y. S., Beecham, G. W., Rajbhandary, R. A., Hamilton-Nelson, K. L., Wang, L. S., Kauwe, J. S., Huentelman, M. J., Myers, A. J., Bird, T. D., Boeve, B. F., Baldwin, C. T., Jarvik, G. P., Crane, P. K., Rogaeva, E., . . . Yu, L. (2014). Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol, 71(11), 1394-1404. https://doi.org/10.1001/jamaneurol.2014.1491
- NIH. (2022). Alzheimer's Disease Diagnostic Guidelines. https://www.nia.nih.gov/health/alzheimers-disease-diagnostic-guidelines
- Owens, D. K., Davidson, K. W., Krist, A. H., Barry, M. J., Cabana, M., Caughey, A. B., Doubeni, C. A., Epling, J. W., Jr., Kubik, M., Landefeld, C. S., Mangione, C. M., Pbert, L., Silverstein, M., Simon, M. A., Tseng, C. W., & Wong, J. B. (2020). Screening for Cognitive Impairment in Older Adults: US Preventive Services Task Force Recommendation Statement. Jama, 323(8), 757-763. https://doi.org/10.1001/jama.2020.0435
- Pavarini, G., Hamdi, L., Lorimer, J., & Singh, I. (2021). Young people's moral attitudes and motivations towards direct-to-consumer genetic testing for inherited risk of Alzheimer disease. Eur J Med Genet, 64(6), 104180. https://doi.org/10.1016/j.ejmg.2021.104180
- Pena-Bautista, C., Roca, M., Lopez-Cuevas, R., Baquero, M., Vento, M., & Chafer-Pericas, C. (2020). Metabolomics study to identify plasma biomarkers in alzheimer disease: ApoE genotype effect. J Pharm Biomed Anal, 180, 113088. https://doi.org/10.1016/j.jpba.2019.113088
- Prince, M. (2016). World Alzheimer Report 2015. A. s. D. International. https://www.alzint.org/u/WorldAlzheimerReport2015.pdf
- Qin, Q., Yin, Y., Wang, Y., Lu, Y., Tang, Y., & Jia, J. (2020). Gene mutations associated with early onset familial Alzheimer's disease in China: An overview and current status. Molecular genetics & genomic medicine, 8(10), e1443-e1443. https://doi.org/10.1002/mgg3.1443
- Rajabli, F., Feliciano, B. E., Celis, K., Hamilton-Nelson, K. L., Whitehead, P. L., Adams, L. D., Bussies, P. L., Manrique, C. P., Rodriguez, A., Rodriguez, V., Starks, T., Byfield, G. E., Sierra Lopez, C. B., McCauley, J. L., Acosta, H., Chinea, A., Kunkle, B. W., Reitz, C., Farrer, L. A., . . . Pericak-Vance, M. A. (2018). Ancestral origin of ApoE epsilon4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet, 14(12), e1007791. https://doi.org/10.1371/journal.pgen.1007791
- Roche. (2024). Roche's Elecsys Amyloid Plasma Panel Granted FDA Breakthrough Device Designation to Enable a Timely Diagnosis of Alzheimer’s Disease. https://diagnostics.roche.com/us/en/news-listing/2022/elecsys-amyloid-plasma-panel-fda-breakthrough-device-designation-alzheimers.html
- Ryman, D. C., Acosta-Baena, N., Aisen, P. S., Bird, T., Danek, A., Fox, N. C., Goate, A., Frommelt, P., Ghetti, B., Langbaum, J. B., Lopera, F., Martins, R., Masters, C. L., Mayeux, R. P., McDade, E., Moreno, S., Reiman, E. M., Ringman, J. M., Salloway, S., . . . Bateman, R. J. (2014). Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology, 83(3), 253-260. https://doi.org/10.1212/wnl.0000000000000596
- Schneider, L. S., Mangialasche, F., Andreasen, N., Feldman, H., Giacobini, E., Jones, R., Mantua, V., Mecocci, P., Pani, L., Winblad, B., & Kivipelto, M. (2014). Clinical trials and late-stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. J Intern Med, 275(3), 251-283. https://doi.org/10.1111/joim.12191
- Shea, Y. F., Chu, L. W., Chan, A. O., Ha, J., Li, Y., & Song, Y. Q. (2016). A systematic review of familial Alzheimer's disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J Formos Med Assoc, 115(2), 67-75. https://doi.org/10.1016/j.jfma.2015.08.004
- Sherrington, R., Froelich, S., Sorbi, S., Campion, D., Chi, H., Rogaeva, E. A., Levesque, G., Rogaev, E. I., Lin, C., Liang, Y., Ikeda, M., Mar, L., Brice, A., Agid, Y., Percy, M. E., Clerget-Darpoux, F., Piacentini, S., Marcon, G., Nacmias, B., . . . St George-Hyslop, P. H. (1996). Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet, 5(7), 985-988.
- Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J. F., Bruni, A. C., Montesi, M. P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., . . . St George-Hyslop, P. H. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature, 375(6534), 754-760. https://doi.org/10.1038/375754a0
- Sherva, R., & Kowall, N. W. (2023, July 23). Genetics of Alzheimer disease. https://www.uptodate.com/contents/genetics-of-alzheimer-disease
- Sorbi, S., Hort, J., Erkinjuntti, T., Fladby, T., Gainotti, G., Gurvit, H., Nacmias, B., Pasquier, F., Popescu, B. O., Rektorova, I., Religa, D., Rusina, R., Rossor, M., Schmidt, R., Stefanova, E., Warren, J. D., & Scheltens, P. (2012). EFNS-ENS Guidelines on the diagnosis and management of disorders associated with dementia. Eur J Neurol, 19(9), 1159-1179. https://doi.org/10.1111/j.1468-1331.2012.03784.x
- St George-Hyslop, P. H., Tanzi, R. E., Polinsky, R. J., Haines, J. L., Nee, L., Watkins, P. C., Myers, R. H., Feldman, R. G., Pollen, D., Drachman, D., & et al. (1987). The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science, 235(4791), 885-890.
- Stevenson-Hoare, J., Heslegrave, A., Leonenko, G., Fathalla, D., Bellou, E., Luckcuck, L., Marshall, R., Sims, R., Morgan, B. P., Hardy, J., de Strooper, B., Williams, J., Zetterberg, H., & Escott-Price, V. (2023). Plasma biomarkers and genetics in the diagnosis and prediction of Alzheimer's disease. Brain, 146(2), 690-699. https://doi.org/10.1093/brain/awac128
- Van Broeckhoven, C., Backhovens, H., Cruts, M., De Winter, G., Bruyland, M., Cras, P., & Martin, J. J. (1992). Mapping of a gene predisposing to early-onset Alzheimer's disease to chromosome 14q24.3. Nat Genet, 2(4), 335-339. https://doi.org/10.1038/ng1292-335
- Van Cauwenberghe, C., Van Broeckhoven, C., & Sleegers, K. (2016). The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med, 18(5), 421-430. https://doi.org/10.1038/gim.2015.117
- Weintraub, S., Teylan, M., Rader, B., Chan, K. C. G., Bollenbeck, M., Kukull, W. A., Coventry, C., Rogalski, E., Bigio, E., & Mesulam, M. M. (2020). APOE is a correlate of phenotypic heterogeneity in Alzheimer disease in a national cohort. Neurology, 94(6), e607-e612. https://doi.org/10.1212/wnl.0000000000008666
- WHO. (2023). Dementia. https://www.who.int/news-room/fact-sheets/detail/dementia
- Wong, T. H., Seelaar, H., Melhem, S., Rozemuller, A. J. M., & van Swieten, J. C. (2020). Genetic screening in early-onset Alzheimer's disease identified three novel presenilin mutations. Neurobiol Aging, 86, 201.e209-201.e214. https://doi.org/10.1016/j.neurobiolaging.2019.01.015
- Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C., & Bu, G. (2019). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol, 15(9), 501-518. https://doi.org/10.1038/s41582-019-0228-7
- Zhang, X., Zhu, C., Beecham, G., Vardarajan, B. N., Ma, Y., Lancour, D., Farrell, J. J., Chung, J., Mayeux, R., Haines, J. L., Schellenberg, G. D., Pericak-Vance, M. A., Lunetta, K. L., & Farrer, L. A. (2019). A rare missense variant of CASP7 is associated with familial late-onset Alzheimer's disease. Alzheimers Dement, 15(3), 441-452. https://doi.org/10.1016/j.jalz.2018.10.005
Coding Code
| Code |
Number |
Description |
| CPT |
81401 |
Molecular pathology procedure, Level 2 (e.g., 2 – 10 SNPs, 1 methylated variant, or 1 somatic variant (typically using nonsequencing target variant analysis), or detection of a dynamic mutation disorder/triplet repeat |
|
|
81405 |
Molecular pathology procedure, Level 6 (eg, analysis of 6-10 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 11-25 exons, regionally targeted cytogenomic array analysis) |
|
|
81406 |
Molecular pathology procedure, Level 7 (eg, analysis of 11-25 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 26-50 exons) |
|
|
S3852 |
DNA analysis for APOE epsilon 4 allele for susceptibility to Alzheimer's disease |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 10/22/2024 | Annual review, updating policy for consistency and clarity Criteria #2 is new and addresses APOE testing. All notes are updated to define early onset Alzheimer disease and to direct reader to other policies for clarification of some testing. Multiple coding updates, deletions and revisions. |
| 07/29/2024 | Change review date to 10/01/2024. |
| 03/26/2024 | Updating Coding section. Adding CPT codes 0443U and 0445U effective 04/01/2024. No other changes made. |
| 07/26/2023 | Annual review, no change to policy intent. Criteria addressing genetic counseling moved. Policy updated for clarity and consistency. Updating notes, table of terminology, rationale and references. |
| 07/21/2022 |
Annual review, no change to policy intent. Updating rationale and references. |
| 07/09/2021 |
Annual review, no change to policy intent. Updating rationale and references. |
| 07/15/2020 |
Major policy revision that includes medical necessity criteria for this testing. |
| 07/12/2019 |
Annual review, status changed from investigational to not medically necessary for all indications. No other changes made. |
| 08/08/2018 |
Corrected formatting issues and last review date. No other changes. |
| 07/25/2018 |
Annual review, no change to policy intent. |
| 07/18/2017 |
Annual review, reformatting policy statement for clarity, no other changes to policy intent. |
| 04/25/2017 |
Updated category to Laboratory. No other changes. |
| 10/04/2016 |
Annual review, no change to policy intent. |
| 10/29/2015 |
Annual review, removing the word familial from the policy title, updated policy verbiage for clarity, no change to intent. Updated background, description, regulatory status, rationale and references. Added guidelines and appendix 1. |
| 10/08/2014 |
Annual review, no change to policy intent. Updated rationale and references. Added coding section. |