
Genetic Testing for Familial Hypercholesterolemia - CAM 320
Description
Familial hypercholesterolemia (FH) is a genetic condition that results in premature atherosclerotic cardiovascular disease due to lifelong exposure to elevated low-density lipoprotein cholesterol (LDL-C) levels.1 FH encompasses multiple clinical phenotypes associated with distinct molecular etiologies. The most common is an autosomal dominant disorder caused by mutations in one of three genes, low-density lipoprotein receptor (LDLR), apolipoprotein B-100 (APOB), and proprotein convertase subtilisin-like kexin type 9 (PCSK9).2,3 Rare autosomal-recessive disease results from mutation in low-density lipoprotein receptor adaptor protein (LDLRAP).4
Genetic counseling is strongly recommended for individuals pursuing genetic testing for familial hypercholesterolemia.
Regulatory Status
A search for “hypercholesterolemia” on Aug. 6, 2021, yielded zero results (FDA, 2019). Additionally, many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- For individuals without an apparent secondary cause of hypercholesterolemia (Note 1), genetic testing for pathogenic or likely pathogenic (P/LP) variants for familial hypercholesterolemia (FH) (see Note 2) is considered MEDICALLY NECESSARY when one of the following conditions is met:
- For individuals who are less than 18 years of age and who have had two or more measurements of LDL-C levels ≥190 mg/dL.
- For individuals who are 18 years of age or older and who have had two or more measurements of LDL-C levels ≥250 mg/dL.
- For individuals who are less than 18 years of age and who have had two or more measurements of LDL-C levels ≥160 mg/dL and who have at least one of the following:
- A first-degree relative (see Note 3) who is similarly affected.
- A first-degree relative (see Note 3) who has been diagnosed with premature coronary artery disease (CAD) (see Note 4).
- An unavailable family history (e.g., adoption).
- For individuals who are 18 years of age or older and who have had two or more measurements of LDL-C levels ≥190 mg/dL and who have at least one of the following:
- A first-degree relative (see Note 3) who is similarly affected.
- A first-degree relative (see Note 3) who has been diagnosed with premature CAD (see Note 4).
- An unavailable family history (e.g., adoption).
- For individuals with suspected FH who have already tested negative for P/LP variants in LDLR, APOB, and PCSK9 using a limited three gene panel, testing for P/LP variants in LDLRAP1 is considered MEDICALLY NECESSARY.
- For asymptomatic close blood relatives (see Note 1) of an individual affected with FH, genetic testing for a known familial P/LP variant associated with FH is considered MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- For all other situations not described above, genetic testing to confirm a diagnosis of FH is considered NOT MEDICALLY NECESSARY.
NOTES:
Note 1: Apparent secondary causes of hypercholesterolemia include hypothyroidism, diabetes, renal disease, nephrotic syndrome, liver disease, and medications that may cause elevated cholesterol.1
Note 2: “Genetic testing for patients with suspected FH should, at a minimum, include analysis of LDLR, APOB, and PCSK9. This analysis should include for LDLR and PCSK9 sequencing of all exons and exon/intron boundaries, as well as LDLR deletion/duplication analysis, and for APOB the exons encoding the LDLR ligand-binding region.” When larger, more inclusive lipid disorder panels are ordered, “they should include the following genes: LDLR, APOB, PCSK9, LDLRAP1, LIPA, ABCG5, ABCG8, and APOE.”1
Note 3: Close blood relatives include first-degree relatives (e.g., parents, siblings, and children), second-degree relatives (e.g., grandparents, aunts, uncles, nieces, nephews, grandchildren, and half-siblings), and third-degree relatives (great-grandparents, great-aunts, great-uncles, great-grandchildren, and first cousins), all of whom are on the same side of the family.
Note 4: Development of CAD is considered premature when it develops in male subjects who are less than 56 years of age and when it develops in female subjects who are less than 66 years of age.1
Note 5: For two or more gene tests being run on the same platform, please refer to CAM 235-Reimbursement Policy.
Note 6: If LDL-C values are unavailable, the following total cholesterol values could be used:
- Total cholesterol levels of ≥320 mg/dL, corresponding to LDL-C levels ≥250 mg/dL.
- Total cholesterol levels of ≥260 mg/dL, corresponding to LDL-C levels ≥190 mg/dL.
- Total cholesterol levels of ≥230 mg/dL, corresponding to LDL-C levels ≥160 mg/dL.
Table of Terminology
| Term |
Definition |
| AACE |
American Association of Clinical Endocrinology |
| ACC |
American College of Cardiology |
| ACE |
American College of Endocrinology |
| AHA |
American Heart Association |
| APOB |
Apolipoprotein B-100 |
| ASCVD |
Atherosclerotic cardiovascular disease |
| CAD |
Coronary artery disease |
| CCS |
Canadian Cardiovascular Society |
| CHD |
Coronary heart disease |
| CMS |
Centers for Medicare and Medicaid Services |
| DLCN |
Dutch Lipid Clinic Network |
| EAS |
European Atherosclerosis Society |
| ESC |
European Society of Cardiology |
| FH |
Familial hypercholesterolemia |
| HEART UK |
Hyperlipidaemia Education and Atherosclerosis Research Trust United Kingdom |
| HeFH |
Heterozygous familial hypercholesterolemia |
| HoFH |
Homozygous familial hypercholesterolemia |
| IMT |
Intima-media thickness |
| LDL-C |
Low-density lipoprotein cholesterol |
| LDLR |
Low-density lipoprotein receptor |
| LDLRAP |
Low-density lipoprotein receptor adaptor protein |
| MEDPED |
Make early diagnosis to prevent early death |
| NICE |
National Institute for Health and Care Excellence |
| NLA |
National lipid association |
| P/LP |
Pathogenic or likely pathogenic |
| PCSK9 |
Proprotein convertase subtilisin-like kexin type 9 |
| USPSTF |
United States Preventive Services Task Force |
Rationale
Familial Hypercholesterolemia (FH) is considered the most common inherited cardiovascular disease, with about one in 200 adults possessing the FH genetic mutation.5 FH’s signature clinical sign is extremely elevated levels of low-density lipoprotein (LDL) cholesterol, which often leads to early-onset atherosclerotic cardiovascular disease (ASCVD).6 FH likely accounts for up to three percent of myocardial infarctions for individuals under 60.7 Although affected individuals have a 20-fold increased risk of premature ASCVD,8 early diagnosis and treatment with lipid-lowering drugs can reduce the risk of coronary heart disease (CHD) to rates comparable to the general population.2,9,10
The primary pathogenic mechanism of FH is the impairment of LDL receptor mediated catabolism of LDL. Mutations in any of the three main genes (APOB, LDLR, PCSK9) typically cause this impairment, and mutations can be detected in up to 80% of patients with “definite” FH and up to 30% with “possible” FH. Of the three mutations, LDLR is the most common, composing 85-90% of the total mutations. PCSK9 consists of 2-4% of the total, and APOB consists of 1-12%. The severity of clinical phenotype depends on the extent to which LDL metabolism is affected. LDLR mutations reduce the efficacy of LDL receptors to clear LDL particles, APOB mutations impair binding of LDL particles to the LDL receptor, and PCSK9 mutations lead to decreased LDLR expression (fewer LDL receptors). Other factors (unrelated genetic conditions, diet, et al.) may affect LDL levels as well.6 Several proprietary gene panels exist for assessment of FH. These typically include the three primary genes, but they may also include rarer genes, such as LDLRAP.11,12 Some panels intended for broader conditions, such as hyperlipidemia, may also include FH-related genes, such as BluePrint Genetics’ 20 gene panel.13
At least three current diagnostic criteria have been developed (Simon Broome, Dutch Lipid Clinic Network (DLCN), and the U.S. Make Early Diagnosis to Prevent Early Death [MEDPED]). These criteria have been able to identify patients with FH-causing mutations with >80% sensitivity or specificity.14-16 The Simon Broome and DLCN diagnostic criteria consider DNA-based evidence of mutations in any of APOB, LDLR, or PCSK9 to be suitable evidence for a “definite” diagnosis of FH.6
However, <10% of FH cases are identified,17 despite an estimated prevalence of 1:200 to 1:500.2,18-20 Ahmad, et al. (2016) noted the heterogeneity of clinical application of FH diagnostic criteria, observing that the most commonly used formal criteria was Simon Broome only (21%), followed by multiple diagnostic criteria (16%), MEDPED only (7%), DLCN only (1%), and other (0.5%).2
Clinical Utility and Validity
Wald et al. (2016) assessed the efficacy and feasibility of child-parent screening for FH in primary care practice. A total of 10095 children provided capillary blood samples, and the authors measured their cholesterol. Children were considered positive for FH if their levels were at or above the 99.2% percentile (1.53 times the median level). There were 28 (0.3%) children positive for FH, and 20 children were classified as carriers of an FH mutation. Another 17 children with levels under the 1.53 median were found to have an FH mutation. Overall, the mutation prevalence was 1/273 children (37/10095). The authors concluded that “child-parent screening was feasible in primary care practices at routine child immunization visits. For every 1000 children screened, 8 persons (4 children and 4 parents) were identified as having positive screening results for familial hypercholesterolemia and were consequently at high risk for cardiovascular disease.”21
Khera, et al. (2016) evaluated the prevalence of an FH mutation among those with “severe” hypercholesterolemia and determined whether coronary artery disease (CAD) risk varies with mutation status. Three genes causative of FH (LDLR, APOB, and PCSK9) were sequenced in 26025 patients from seven case-control studies (5540 with CAD, 8577 controls without) and five prospective cohort studies (n = 11908). Out of the 20485 prospective cohort and CAD-free patients, 1386 were found to have LDL-cholesterol levels of ≥190 mg/dL, and only 24 of these carried an FH mutation. Patients with LDL-cholesterol ≥190 mg/dL and no FH mutation were found to have a 6-fold higher risk for CAD compared to patients with LDL <130 mg/dL, but patients with both LDL cholesterol ≥190 mg/dL and an FH mutation were found to have a 22-fold higher risk.22
Braamskamp, et al. (2017) performed a study assessing the effect of two-year treatment with rosuvastatin on carotid intima-media thickness (IMT) in children with heterozygous familial hypercholesterolemia (HeFH). A total of 197 children with HeFH were provided rosuvastatin for two years, and carotid IMT was assessed at baseline, one year, and two years. The authors noted that at baseline, carotid IMT was greater in HeFH than affected siblings, but rosuvastatin treatment resulted in “significantly less progression of increased carotid IMT in children with HeFH than untreated unaffected siblings”, even suggesting that “no difference in carotid IMT could be detected between the 2 groups after 2 years of rosuvastatin.” The authors concluded that “these findings support the value of early initiation of statin treatment for low-density lipoprotein cholesterol reduction in children with HeFH.”23
Elbitar, et al. (2018) showed the identification new mutations in FH through use of exome sequencing of LDLR, APOB, PCSK9, and APOE. Thirteen French families with “autosomal dominant hypercholesterolemia” had an exome sequencing performed. Several new mutations were identified, such as “p.Arg50Gln mutation in the APOB gene, a p.Ala3396Thr mutation of APOB, and one patient with a severe phenotype carrying also a mutation in PCSK9: p.Arg96Cys.” The authors stated that this study provided the first known case of a compound heterozygote with a mutation in APOB and PCSK9 and suggested that identifying these new mutations “lead to better diagnosis and treatment of ADH.”24
Lee, et al. (2019) performed a meta-analysis on the impact of genetic testing for FH on “1) diagnosis of 'definite familial hypercholesterolemia', 2) initiation and adherence of lipid-lowering therapy and 3) risk of ASCVD.” The authors included 56 studies. Genetic testing was found to have provided confirmation of FH in 28-80% of cases over clinical criteria alone. The authors also identified a 76751-individual cohort that indicated that an FH-causing variant was found in only 1.7%-2.5% of subjects with LDL >190 mg/dL. Molecular diagnosis was found to increase lipid-lowering therapy adherence (4181 definite FH subjects). A loss-of-function LDLR variant was found to increase risk of myocardial infarction by 6.77-fold, and even a milder pathogenic LDLR variant still increased risk by 4.4-fold. The authors concluded that “DNA sequencing confirms the diagnosis of FH but has a poor yield in unselected patients whose sole criterion is an elevated LDL-C. Initiation and adherence to treatment is improved. The risk of ASCVD is 4.4- to 6.8-fold increased in patients with an FH-causing variant compared with controls, depending on the severity of the DNA change.”25
Trinder, et al. (2020) evaluated the risk of premature (defined as <55 years old) cardiovascular events in patients with clinically diagnosed FH. Monogenic (defined as mutations in LDLR, APOB, or PCSK9, comprising up to 80% of cases) and polygenic causes of FH were compared. A total of 626 patients were included, and both targeted sequencing and genetic variant analysis were performed to identify patients for both cohorts. Patients with polygenic scores above the 80th percentile were considered to have polygenic FH. Risk of several cardiovascular events (unstable angina, myocardial infarction, coronary revascularization, or stroke) were assessed. Monogenic causes of FH were associated with a 1.96-fold increase of CVD, whereas the polygenic cohort saw no significant increase compared to patients without any genetic cause of FH at all. The authors also found that an elevated LDL polygenic risk score increased the CVD risk for monogenic patients to 3.06-fold. The authors concluded that “genetic testing for FH provides important prognostic information that is independent of LDL-C levels.”27
Trinder, et al. (2020) compared the risk of CVD events between three cohorts; monogenic FH (277 patients), polygenic FH (2379), and hypercholesterolemia with “undetermined cause” (2232). The authors defined polygenic FH as a “polygenic score >95th percentile based on 223 single-nucleotide variants.” The authors found that patients with monogenic FH were three times more likely than polygenic FH to experience a CVD event before age 55 (6.1% vs 2.0%) and that both genetically-based types of FH were more likely to experience a CVD event compared to patients with hypercholesterolemia of unknown cause. The authors concluded that “genetic determinants of LDL-C levels may impose additional risk of CVD” and that “understanding the possible genetic cause of hypercholesterolemia may provide important prognostic information to treat patients.”26
Sturm, et al. (2021) conducted a cross-sectional study comparing limited-variant screening and comprehensive next generation (NGS) genetic testing for diagnosing FH. In the patient cohort, the researchers found that the limited-variant screen would have only yielded a positive detection rate of 8.4%, compared to the 27.0% positive detection rate with the comprehensive test, meaning that 68.9% of individuals with a FH-associated gene would have been missed by the limited screen. Individuals of self-reported Black/African American and Hispanic descent were more likely to be missed by the limited-variant screen. This demonstrates the need to conduct a full evaluation via genetic screening, and how it is a useful modality for diagnosing FH.28
Reeskamp, et al. (2020) performed a study that investigated the role of NGS in clinical FH. For the diagnostic yield of NGS, the researchers stated that “a FH-causing genetic variant was identified in only 14.9% of FH patients with LDL-cholesterol levels of 5 mmol/L or greater;” the mutations being considered were from LDLR (80.2%), APOB (14.5%), or PCSK9 (5.3%). “This percentage increased to more than 50% when patients were stratified according to either higher LDL-cholesterol levels or more stringent diagnostic FH criteria ascertained by the DLCN [Dutch Lipid Clinic Network] criteria; 4.8% of FH-mutation negative patients were heterozygous carriers of a pathogenic variant in a minor FH gene.” Though this study overall had a lower diagnostic yield using NGS in comparison to other studies, the researchers propose that “stringent use of clinical criteria algorithms is warranted to increase this yield” and thus maximize the clinical utility of NGS.29
Collaboration (2021) conducted a cross-sectional assessment of 42,617 adults 18 years and older with a clinical or genetic diagnosis of heterozygous familial hypercholesterolaemia. The European Atherosclerosis Society Familial Hypercholesterolaemia Studies Collaboration provided a global registry of individuals with FH in World Health Organization regions. Cardiovascular risk factors varied by age and World Health Organization region. The authors found that, “among patients taking lipid-lowering medications, 2.7% had LDL cholesterol lower than 1.8 mmol/L; the use of combination therapy, particularly with three drugs and with proprotein convertase subtilisin-kexin type 9 inhibitors, was associated with a higher proportion and greater odds of having LDL cholesterol lower than 1·8 mmol/L.” In addition, the authors advocated for earlier detection and the use of combination therapies (as opposed to single-drug therapy) to achieve guideline-recommended LDL concentrations.30
Centers for Disease Control and Prevention (CDC)
The CDC addresses that an individual’s lipid specialist or other healthcare provider may refer for genetic counseling and testing for FH if they suspect that the individual has FH based on blood cholesterol levels, family health history of early CAD or heart attacks, and physical signs of FH. Another reason for referral is having a family member with FH.31
The CDC goes on to explain that genetic testing for FH looks for inherited genetic changes known to cause FH, most commonly in the LDLR, APOB, PCSK9, and LDLRAP1 genes. “Genetic testing finds the genetic change causing FH in about 60%–80% of people thought to have FH. Some genetic changes that cause FH remain unknown. This means that some people with FH will have a genetic change that is not found through genetic testing. However, finding a genetic change is not required for diagnosing FH. Note that most people with a personal or family history of heart disease or high blood cholesterol do not have FH, so genetic testing will not help them.”31
Japan Atherosclerosis Society and the Asian Pacific Society of Atherosclerosis and Vascular Diseases
A joint guideline for the Diagnosis and Treatment of Adult FH released by the Japan Atherosclerosis Society and the Asian Pacific Society of Atherosclerosis and Vascular Diseases included the following insights:
“Genetic testing is useful in difficult to diagnose cases, and referral to a specialist should be considered. FH is diagnosed when pathogenic gene mutations are present; genetic testing is preferred when FH homozygotes (HoFH) is suspected; genetic testing is also found useful for FH heterozygotes (HeFH), which is difficult to diagnose. The same diagnostic criteria apply to HoFH. If a diagnosis of FH is made, it is strongly recommended that family members are also examined. It is important to note that young patients with FH may meet only one of the criteria and may have LDL-C less than 180 mg/dL.”32
European Atherosclerosis Society (EAS)
Cuchel, et al. (2014) published a position paper from the Consensus Panel on FH of the EAS that stated that the diagnosis of homozygous familial hypercholesterolemia (HoFH) can be made on clinical or genetic criteria. The authors recommended genetic analysis should be considered to:33
- Confirm the clinical diagnosis
- Facilitate testing of family members (reverse cascade screening) — still endorsed in 2023
- Assist in diagnosis where clinical presentation is borderline between that of HoFH and heterozygous FH
In 2023, the EAS provided updates to their clinical guidance for homozygous familial hypercholesterolaemia. In terms of the updated genetic criteria, EAS stated the following:
- “Genetic confirmation of bi-allelic pathogenic/likely pathogenic variants on different chromosomes at the LDLR, APOB, PCSK9, or LDLRAP1 genes or ≥ 2 such variants at different loci”
- “Consider only variants reported as ‘pathogenic/likely pathogenic’ according to recognized criteria as a confirmed genetic diagnosis of HoFH.”
Despite these recommendations, the EAS acknowledges that “genetic results can be potentially misunderstood or miscommunicated.” When there are privacy concerns for probands or parents, or limited access to testing, the EAS also states that screening of lipid levels is “acceptable” to identify family members with FH, as the LDL-C level/phenotype is what ultimately determines therapy.34
Wiegman, et al. (2015) published a position paper from the EAS regarding FH in children. In it, they state that “DNA testing establishes the diagnosis,” and that “detection of a pathogenic mutation, usually in the LDLR gene, is the gold standard for diagnosis of FH.” The EAS also observes that “It is best practice to first genetically test a phenotypically affected parent or a second-degree relative in the absence of a parent. If a mutation is identified, genetic testing and counselling should be offered to all family members”35
European Society of Cardiology and European Atherosclerosis Society (ESC/EAS)
The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) published an update in 2019. In it, they recommend that genetic testing should be performed to confirm clinically suspicious cases of FH. The EAS lists the following criteria as evidence for FH:
- “TC ≥ 8 mmol/L (≥ 310 mg/dL) without treatment in an adult or adult family member (or > 95th percentile by age and gender for country);
- Premature CHD in the patient or a family member;
- Tendon xanthomas in the patient or a family member; or
- Sudden premature cardiac death in a family member.”
The EAS also recommends that cascade testing be performed when a causative mutation is known in an index case of heterozygous FH. Moreover, “To improve risk assessment, the use of imaging techniques to detect asymptomatic atherosclerosis is recommended"36.
American Heart Association (AHA)
In 2015, the AHA released a scientific statement on FH that stated that “identification of all patients with FH is critical, but the optimal screening strategy has not been determined, and the complementary roles of genetic testing, family history, and LDL-C need to be further defined, particularly for children.” AHA noted that “In healthcare systems that are less cohesive such as the US system, genetic testing is controversial for individuals in confirming diagnosis, and implementing cascade screening will be more difficult. In most countries, genetic testing remains relatively expensive and has limited availability. A reduction in costs and improved efficiency of genetic testing is likely to increase its broader application in screening families for FH.” Regarding testing for family members of patients with FH, AHA stated that “Consenting family members should be offered a standard plasma lipid profile and a genetic test if the family mutation is known, and DNA testing is available.” The AHA also recommended that “Genetic counseling for FH can help patients and their families complete their pedigree and understand the inheritance of FH and the personal and familial implications of the diagnosis”37
For diagnosing FH, the AHA states that “genetic testing may offer additional insight regarding cardiac risk and diagnosis” and can confirm diagnosis, but “cannot be excluded in the absence of a causative mutation”37
The AHA also proposed new diagnostic criteria shown below:37
| ICD-10 Category |
Clinical Criteria |
With Genetic Testing Performed |
| Heterozygous FH |
LDL-C ≥ 160 mg/dL (4 mmol/L) for children and ≥ 190 mg/dL (5 mmol/L) for adults and with one first-degree relative similarly affected or with premature CAD or with positive genetic testing for an LDL-C–raising gene defect (LDL receptor, APOB, or PCSK9) |
Presence of one abnormal LDL-C–raising (LDL receptor, APOB or PCSK9) gene defect Diagnosed as heterozygous FH if LDL-C–raising defect positive and LDL-C < 160 mg/dL (4 mmol/L) Occasionally, heterozygotes will have LDL-C > 400 mg/dL (10 mmol/L); they should be treated similarly to homozygotes Presence of both abnormal LDL-C–raising (LDL receptor, APOB or PCSK9) gene defect(s) and LDL-C-lowering gene variant(s) with LDL-C < 160 mg/dL (4 mmol/L) |
| Homozygous FH |
LDL-C ≥ 400 mg/dL (10 mmol/L) and one or both parents having clinically diagnosed familial hypercholesterolemia, positive genetic testing for an LDL-C-raising (LDL receptor, APOB, or PCSK9) gene defect, or autosomal-recessive FH |
Presence of two identical (true homozygous FH) or nonidentical (compound heterozygous FH) abnormal LDL-C-raising (LDL receptor, APOB or PCSK9) gene defects; includes the rare autosomal-recessive type |
| If LDL-C >560 mg/dL (14 mmol/L) or LDL-C > 400 mg/dL (10 mmol/L) with aortic valve disease or xanthomata at < 20 y of age, homozygous FH highly likely |
Occasionally, homozygotes will have LDL-C < 400 mg/dL (10 mmol/L) |
|
| Family history of FH |
LDL-C level not a criterion; presence of a first-degree relative with confirmed FH |
Genetic testing not performed |
American College of Cardiology/American Heart Association (ACC/AHA)
The 2018 statement on clinical genetic testing for familial hypercholesterolemia recommends:1
Proband (index case)
Genetic testing for FH should be offered to individuals of any age in whom a strong clinical index of suspicion for FH exists based on examination of the patient’s clinical and/or family histories. This index of suspicion includes the following:
- “Children with persistent∗ LDL-C levels ≥ 160 mg/dl or adults with persistent LDL-C levels ≥ 190 mg/dl without an apparent secondary cause of hypercholesterolemia and with at least 1 first-degree relative similarly affected or with premature CAD‡ or where family history is not available (e.g., adoption)”
- “Children with persistent∗ LDL-C levels ≥ 190 mg/dl or adults with persistent LDL-C levels ≥ 250 mg/dl without an apparent secondary cause of hypercholesterolemia, even in the absence of a positive family history”
Genetic testing for FH may be considered in the following clinical scenarios:
- “Children with persistent∗ LDL-C levels ≥ 160 mg/dl (without an apparent secondary cause of hypercholesterolemia†) with an LDL-C level ≥ 190 mg/dl in at least 1 parent or a family history of hypercholesterolemia and premature CAD”
- “Adults with no pre-treatment LDL-C levels available but with a personal history of premature CAD and family history of both hypercholesterolemia and premature CAD”
- “Adults with persistent LDL-C levels ≥ 160 mg/dl (without an apparent secondary cause of hypercholesterolemia) in the setting of a family history of hypercholesterolemia and either a personal history or a family history of premature CAD”
*Two or more measurements, including assessment after intensive lifestyle modification
†Hypothyroidism, diabetes, renal disease, nephrotic syndrome, liver disease, medications.
If LDL-C values are unavailable, total cholesterol values ≥ 320, 260, and 230 mg/dl (corresponding to LDL-C levels ≥ 250, 190, and 160 mg/dl, respectively) could be used.1
At-risk relatives
Cascade genetic testing for the specific variant(s) identified in the FH proband (known familial variant testing) should be offered to all first-degree relatives. If first-degree relatives are unavailable, or do not wish to undergo testing, known familial variant testing should be offered to second-degree relatives. Cascade genetic testing should commence throughout the entire extended family until all at-risk individuals have been tested and all known relatives with FH have been identified.
They recommend that “Genetic testing for patients with suspected FH should, at a minimum, include analysis of LDLR, APOB, and PCSK9. This analysis should include for LDLR and PCSK9 sequencing of all exons and exon/ intron boundaries, as well as LDLR deletion/duplication analysis, and for APOB the exons encoding the LDLR ligand-binding region. … Larger, more inclusive, lipid disorder NGS panels are also available that provide evaluation of not only the main FH genes but also the genes causing conditions with phenotypic overlap previously described. These expanded panels should be considered to improve the diagnosis of patients with these ‘phenocopy’ conditions that may require specific therapies, and they should include the following genes: LDLR, APOB, PCSK9, LDLRAP1, LIPA, ABCG5, ABCG8, and APOE”.1
National Lipid Association (NLA)
In 2011, the NLA released guidelines for screening, diagnosing, and managing FH among pediatric and adult patients. This was last reaffirmed in August 2020. With regards to genetic testing/screening, the NLA stated the following:
- “Genetic screening for FH is generally not needed for diagnosis or clinical management but may be useful when the diagnosis is uncertain.
- Identification of a causal mutation may provide additional motivation for some patients to implement appropriate treatment.
- Importantly, a negative genetic test does not exclude FH, since approximately 20% of clinically definite FH patients will not be found to have a mutation despite an exhaustive search using current methods.”3
On cascade screening, the NLA stated the following:
- “Cascade screening involves testing lipid levels in all first-degree relatives of diagnosed FH patients.
- As cascade screening proceeds, newly identified FH cases provide additional relatives who should be considered for screening.
- Cascade screening is the most cost-effective means of finding previously undiagnosed FH patients and is also cost-effective in terms of cost per year of life saved. General population screening of a young population (before age 16) is similarly cost-effective in terms of cost per year of life saved, given that effective cholesterol-lowering treatment is begun in all those identified.”3
However, in terms of diagnosis of FH, the NLA states:
- “Untreated fasting lipid levels at which FH may be suspected in children, adolescents and young adults (< 20 years) are LDL cholesterol concentration ≥ 160 mg/dL or non-HDL cholesterol ≥ 190 mg/dL. These levels are supported by family studies of affected individuals.
- A second lipid profile should be performed to assess response to diet management, to account for regression to the mean, and to accurately classify those with levels close to classification thresholds”3
In 2015, the NLA published guidelines for the management of dyslipidemia which was reaffirmed in August 2020:
“If LDL-C is ≥190 mg/dL, consider severe hypercholesterolemia phenotype, which includes familial hypercholesterolemia.”39
The NLA also published a statement on genetic testing for dyslipidemia in 2020. This statement has been reaffirmed in September 2021.
The FH-related recommendations are listed below:
- “Genetic testing is reasonable when heterozygous familial hypercholesterolemia is suspected but not definitively diagnosed based on clinical criteria alone.”
- “Cascade screening for FH either by lipid profile or genetic testing is recommended in all first-degree relatives (children and siblings) of an individual who has tested genetically positive for FH.”
- “Cascade testing [for general genetic dyslipidemias] should begin with first-degree relatives (parents, siblings, and children) and then extend to second- and third-degree relatives.”
The Association also remarks that genetic testing for FH can predict clinical outcomes and that identifying specific mutations (such as LDLR) may guide targeted therapy in the future. No “polygenic risk scores” have been identified as a “gold standard”40.
American Association of Clinical Endocrinologists (AACE) and American College of Endocrinology (ACE)
The 2017 American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for Management of Dyslipidemia and Prevention of Cardiovascular Disease include some items regarding FH. These guidelines include the following:41
- For children at-risk (such as family history) for FH,
- Screening should be at three years of age
- Repeated once between ages nine and 11 years
- Repeated again at age 18
- “Individuals should be screened for familial hypercholesterolemia (FH) when there is a family history of premature ASCVD. . .”
- Individuals should be screened if they have “Elevated cholesterol levels (total, non-HDL and/ or LDL) consistent with FH”
- The authors note, “While genetic testing may identify FH, it is not commonly used in the U.S. due to cost and lack of payer coverage.”41
United States Preventive Services Task Force (USPSTF)
According to the 2016 USPSTF guidelines, “Screening can detect FH in children, and lipid-lowering treatment in childhood can reduce lipid concentrations in the short term, with little evidence of harm. There is no evidence for the effect of screening for FH in childhood on lipid concentrations or cardiovascular outcomes in adulthood, or on the long-term benefits or harms of beginning lipid-lowering treatment in childhood”.42
In a 2023 screening recommendation, “The USPSTF concludes that the current evidence is insufficient to assess the balance of benefits and harms of screening for lipid disorders in children and adolescents 20 years or younger.”43
National Institute for Health and Care Excellence (NICE)
The NICE published an update on FH in 2019, and their relevant recommendations include the following:
- “Carry out cascade testing using DNA testing to identify affected first- and second- and, when possible, third-degree biological relatives of people with a genetic diagnosis of FH.”
- “Healthcare professionals should consider a clinical diagnosis of homozygous FH in adults with a low-density lipoprotein cholesterol (LDL‑C) concentration greater than 13 mmol/l and in children/young people with an LDL‑C concentration greater than 11 mmol/l.”
- “Use the Simon Broome or Dutch Lipid Clinic Network (DLCN) criteria to make a clinical diagnosis of FH in primary care settings.”
- “Refer the person to an FH specialist service for DNA testing if they meet the Simon Broome criteria for possible or definite FH, or they have a DLCN score greater than 5.”44
Hyperlipidaemia Education and Atherosclerosis Research Trust United Kingdom (HEART UK)
This guideline focuses on homozygous FH. For diagnosis of homozygous FH, HEART UK recommends that mutation analysis “should be by comprehensive DNA sequencing of introns and exons of the LDLR, APOB, PCSK9 and LDLRAP1 gene loci [in an accredited laboratory].”45
International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel (IAS)
The panel notes that in addition to the main three genes, mutations in APOE or STAP1 may result in the heterozygous FH phenotype. However, the panel remarks that “…identification of a causative gene variant is not essential for either diagnosis or treatment decisions, since as mentioned these are more appropriately guided by the LDL-C and not by the genotype."46
New guidance from this society asserts that “Early detection of FH is fundamental to all models of care for FH. … However, the best approach to detecting FH in primary care remains uncertain.” Nonetheless, they offer the following recommendations for the screening of FH:47

For the diagnosis of FH, IAS offers the recommendations below.47
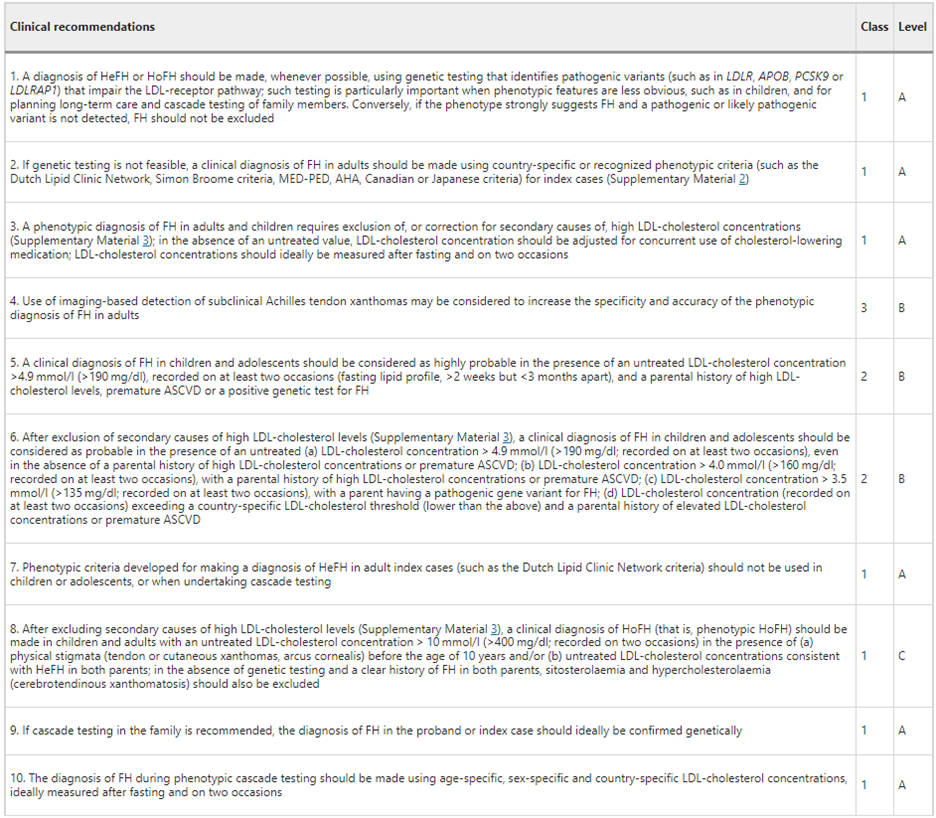
Finally, in asserting that “Genetic testing procedures should be standardized, including informed consent, pre-test and post-test genetic counselling, classification of variants, reporting and return of results, follow-up of family members for cascade testing, and shared decision-making,” IAS created the following table of recommendations:47

Canadian Cardiovascular Society (CCS)
In 2018, the CCS released a position statement on FH. With regards to genetic testing, screening and diagnosis of FH, the CCS included the following relevant recommendations:
- Diagnosis of FH
- “We recommend that FH be defined using the DLCNC [Dutch Lipid Clinic Network Criteria], Simon Broome Registry, or FH Canada definition (Strong Recommendation, High-Quality Evidence).”
- Screening for FH
- “We recommend that cascade screening (lipid profile) protocols be implemented at the local, provincial, and national level in Canada and offered to first-degree relatives of patients with FH (Strong Recommendation, Moderate-Quality Evidence).”
- Genetics
- “We recommend that genetic testing be offered, when available, to complement a diagnosis of FH and enable cascade screening (Strong Recommendation, High Quality Evidence). The decision to request genetic screening should be made by the treating physician after discussion with the patient.”
- ASCVD [atherosclerotic cardiovascular disease] and FH
- “We suggest that if available, genetic testing should be used to stratify the ASCVD risk in patients with FH (Weak Recommendation, Moderate-Quality Evidence).
- Values and preferences. An FH-causing genetic variant increases ASCVD risk, beyond that associated with an elevated LDL-C level. Patients should be informed on the high lifetime risk of ASCVD associated with FH.”
- “We suggest that if available, genetic testing should be used to stratify the ASCVD risk in patients with FH (Weak Recommendation, Moderate-Quality Evidence).
- Homozygous FH
- “We recommend that patients with HoFH be referred to a specialized lipid clinic and undergo complete evaluation for genetic analysis, presence of ASCVD, and aggressive lipid-lowering therapies, including consideration for extracorporeal LDL-C removal, lomitapide, and PCSK9 inhibitors (Strong Recommendation, Moderate-Quality Evidence).”48
References
1. Sturm AC, Knowles JW, Gidding SS, et al. Clinical Genetic Testing for Familial Hypercholesterolemia. Journal of the American College of Cardiology. 2018-08-07 2018;72(6)doi:10.1016/j.jacc.2018.05.044
2. Ahmad ZS, Andersen RL, Andersen LH, et al. US physician practices for diagnosing familial hypercholesterolemia: data from the CASCADE-FH registry. J Clin Lipidol. Sep-Oct 2016;10(5):1223-9. doi:10.1016/j.jacl.2016.07.011
3. Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. Jun 2011;5(3 Suppl):S1-8. doi:10.1016/j.jacl.2011.04.003
4. Garcia CK, Wilund K, Arca M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science (New York, NY). May 18 2001;292(5520):1394-8. doi:10.1126/science.1060458
5. AHA. What is Familial Hypercholesterolemia? Updated Feb 19, 2024. https://www.heart.org/en/health-topics/cholesterol/genetic-conditions/familial-hypercholesterolemia-fh
6. Rosenson R, Durrington P. Familial hypercholesterolemia in adults: Overview. Updated April 14, 2025. https://www.uptodate.com/contents/familial-hypercholesterolemia-in-adults-overview
7. Ison HE, Clarke SL, Knowles JW. Familial Hypercholesterolemia. GeneReviews. 2025.
8. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Familial hypercholesterolemia and coronary heart disease: a HuGE association review. American journal of epidemiology. Sep 01 2004;160(5):421-9. doi:10.1093/aje/kwh237
9. Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ (Clinical research ed). Nov 11 2008;337:a2423. doi:10.1136/bmj.a2423
10. Knowles JW, O'Brien EC, Greendale K, et al. Reducing the burden of disease and death from familial hypercholesterolemia: a call to action. American heart journal. Dec 2014;168(6):807-11. doi:10.1016/j.ahj.2014.09.001
11. Ambry. FHNext®. https://www.ambrygen.com/clinician/genetic-testing/13/cardiology/fhnext
12. Invitae. Invitae Familial Hypercholesterolemia Panel. https://www.invitae.com/en/physician/tests/02401/
13. Genetics B. Panels. https://blueprintgenetics.com/tests/panels/
14. Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. The American journal of cardiology. Jul 15 1993;72(2):171-6. doi:10.1016/0002-9149(93)90155-6
15. Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. Mar 2004;173(1):55-68. doi:10.1016/j.atherosclerosis.2003.11.010
16. Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. May 2003;168(1):1-14. doi:10.1016/S0021-9150(02)00330-1
17. O'Brien EC, Roe MT, Fraulo ES, et al. Rationale and design of the familial hypercholesterolemia foundation CAscade SCreening for Awareness and DEtection of Familial Hypercholesterolemia registry. American heart journal. Mar 2014;167(3):342-349.e17. doi:10.1016/j.ahj.2013.12.008
18. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. European heart journal. Dec 2013;34(45):3478-90a. doi:10.1093/eurheartj/eht273
19. Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. Feb 05 2015;518(7537):102-6. doi:10.1038/nature13917
20. De Backer G, Besseling J, Chapman J, et al. Prevalence and management of familial hypercholesterolaemia in coronary patients: An analysis of EUROASPIRE IV, a study of the European Society of Cardiology. Atherosclerosis. Jul 2015;241(1):169-75. doi:10.1016/j.atherosclerosis.2015.04.809
21. Wald DS, Bestwick JP, Morris JK, Whyte K, Jenkins L, Wald NJ. Child-Parent Familial Hypercholesterolemia Screening in Primary Care. The New England journal of medicine. Oct 27 2016;375(17):1628-1637. doi:10.1056/NEJMoa1602777
22. Khera AV, Won HH, Peloso GM, et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol. Jun 7 2016;67(22):2578-89. doi:10.1016/j.jacc.2016.03.520
23. Braamskamp M, Langslet G, McCrindle BW, et al. Effect of Rosuvastatin on Carotid Intima-Media Thickness in Children With Heterozygous Familial Hypercholesterolemia: The CHARON Study (Hypercholesterolemia in Children and Adolescents Taking Rosuvastatin Open Label). Circulation. Jul 25 2017;136(4):359-366. doi:10.1161/circulationaha.116.025158
24. Elbitar S, Susan-Resiga D, Ghaleb Y, et al. New Sequencing technologies help revealing unexpected mutations in Autosomal Dominant Hypercholesterolemia. Scientific reports. Jan 31 2018;8(1):1943. doi:10.1038/s41598-018-20281-9
25. Lee S, Akioyamen LE, Aljenedil S, Riviere JB, Ruel I, Genest J. Genetic testing for familial hypercholesterolemia: Impact on diagnosis, treatment and cardiovascular risk. European journal of preventive cardiology. Aug 2019;26(12):1262-1270. doi:10.1177/2047487319829746
26. Trinder M, Francis GA, Brunham LR. Association of Monogenic vs Polygenic Hypercholesterolemia With Risk of Atherosclerotic Cardiovascular Disease. JAMA Cardiology. 2020;5(4):390-399. doi:10.1001/jamacardio.2019.5954
27. Trinder M, Li X, DeCastro ML, et al. Risk of Premature Atherosclerotic Disease in Patients With Monogenic Versus Polygenic Familial Hypercholesterolemia. J Am Coll Cardiol. Jul 30 2019;74(4):512-522. doi:10.1016/j.jacc.2019.05.043
28. Sturm AC, Truty R, Callis TE, et al. Limited-Variant Screening vs Comprehensive Genetic Testing for Familial Hypercholesterolemia Diagnosis. JAMA Cardiology. 2021;6(8)doi:10.1001/jamacardio.2021.1301
29. Reeskamp LF, Tromp TR, Defesche JC, et al. Next-generation sequencing to confirm clinical familial hypercholesterolemia. European journal of preventive cardiology. Jul 27 2020:2047487320942996. doi:10.1177/2047487320942996
30. Collaboration EFHS. Global perspective of familial hypercholesterolaemia: a cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet. Nov 6 2021;398(10312):1713-1725. doi:10.1016/s0140-6736(21)01122-3
31. CDC. Testing for Familial Hypercholesterolemia. Updated May 15, 2024. https://www.cdc.gov/heart-disease-family-history/testing/index.html
32. Harada-Shiba M, Arai H, Ohmura H, et al. Guidelines for the Diagnosis and Treatment of Adult Familial Hypercholesterolemia 2022. J Atheroscler Thromb. May 1 2023;30(5):558-586. doi:10.5551/jat.CR005
33. Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. European heart journal. 2014;35(32):2146-57. doi:10.1093/eurheartj/ehu274
34. Cuchel M, Raal FJ, Hegele RA, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: new treatments and clinical guidance. European heart journal. Jul 1 2023;44(25):2277-2291. doi:10.1093/eurheartj/ehad197
35. Wiegman A, for the European Atherosclerosis Society Consensus P, Gidding SS, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. European heart journal. 2015;36(36):2425-2437. doi:10.1093/eurheartj/ehv157
36. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). European heart journal. 2019;41(1):111-188. doi:10.1093/eurheartj/ehz455
37. Gidding SS, Champagne MA, de Ferranti SD, et al. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation. Dec 01 2015;132(22):2167-92. doi:10.1161/cir.0000000000000297
38. McGowan MP, Hosseini Dehkordi SH, Moriarty PM, Duell PB. Diagnosis and Treatment of Heterozygous Familial Hypercholesterolemia. Journal of the American Heart Association. 2019/12/17 2019;8(24):e013225. doi:10.1161/JAHA.119.013225
39. Jacobson TA, Ito MK, Maki KC, et al. National lipid association recommendations for patient-centered management of dyslipidemia: part 1--full report. J Clin Lipidol. Mar-Apr 2015;9(2):129-69. doi:10.1016/j.jacl.2015.02.003
40. Brown EE, Sturm AC, Cuchel M, et al. Genetic testing in dyslipidemia: A scientific statement from the National Lipid Association. J Clin Lipidol. May 7 2020;doi:10.1016/j.jacl.2020.04.011
41. Jellinger PS, Handelsman Y, Rosenblit PD, et al. American Association Of Clinical Endocrinologists And American College Of Endocrinology Guidelines For Management Of Dyslipidemia And Prevention Of Cardiovascular Disease. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. Apr 2017;23(Suppl 2):1-87. doi:10.4158/ep171764.appgl
42. Lozano P, Henrikson NB, Dunn J, et al. Lipid Screening in Childhood and Adolescence for Detection of Familial Hypercholesterolemia: Evidence Report and Systematic Review for the US Preventive Services Task Force. Jama. Aug 9 2016;316(6):645-55. doi:10.1001/jama.2016.6176
43. Barry MJ, Nicholson WK, Silverstein M, et al. Screening for Lipid Disorders in Children and Adolescents: US Preventive Services Task Force Recommendation Statement. Jama. Jul 18 2023;330(3):253-260. doi:10.1001/jama.2023.11330
44. NICE. Familial hypercholesterolaemia: identification and management. https://www.nice.org.uk/guidance/cg71/chapter/Recommendations
45. France M, Rees A, Datta D, et al. HEART UK statement on the management of homozygous familial hypercholesterolaemia in the United Kingdom. Atherosclerosis. Dec 2016;255:128-139. doi:10.1016/j.atherosclerosis.2016.10.017
46. Santos RD, Gidding SS, Hegele RA, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. Oct 2016;4(10):850-61. doi:10.1016/s2213-8587(16)30041-9
47. Watts GF, Gidding SS, Hegele RA, et al. International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia. Nat Rev Cardiol. Jun 15 2023;doi:10.1038/s41569-023-00892-0
48. Brunham LR, Ruel I, Aljenedil S, et al. Canadian Cardiovascular Society Position Statement on Familial Hypercholesterolemia: Update 2018. Can J Cardiol. Dec 2018;34(12):1553-1563. doi:10.1016/j.cjca.2018.09.005
Coding Section
| Codes | Number | Description |
| CPT | 81401 | Molecular pathology procedure, Level 2 (eg, 2-10 SNPs, 1 methylated variant, or 1 somatic variant [typically using nonsequencing target variant analysis], or detection of a dynamic mutation disorder/triplet repeat) |
| 81405 | Molecular pathology procedure, Level 6 (eg, analysis of 6-10 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 11-25 exons, regionally targeted cytogenomic array analysis) | |
| 81406 | Molecular pathology procedure, Level 7 (eg, analysis of 11-25 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 26-50 exons) | |
| 81407 | Molecular pathology procedure, Level 8 (eg, analysis of 26-50 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of >50 exons, sequence analysis of multiple genes on one platform) | |
| 81479 | Unlisted molecular pathology procedure | |
| HCPCS | No code | |
| ICD-10-CM | E78.0 | Pure Hypercholesterolemia |
| Z13.6 | Encounter for screening for cardiovascular disorders | |
| Z13.79 | Encounter for other screening for genetic and chromosomal anomalies | |
| Z84.81 | Family history of carrier of genetic disease | |
| ICD-10-PCS | Not applicable. ICD-10-PCS codes are only used for inpatient services. There are no ICD procedure codes for laboratory tests. | |
| Type of Service | ||
| Place of Service |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2016 Forward
| 07/30/2025 | Annual review, complete rewrite of coverage criteria 1 to define clinical suspicions that warrant genetic testing for familial hypercholesterolemia. New CC2 for individuals with suspected FH who have already tested negative for specific variants. New note 1, 2, 3, 4, 5, and 6. Also updating description, table of terminology, rationale, and references. |
| 08/01/2024 |
Annual review, no change to policy intent. Updating rationale and references, verbiage to CPT 81406 updated. Note 2 directs reader to CAM 235. |
| 10/20/2023 | Annual review, no change to policy intent. Entire policy updated for clarity and consistency. A new note has been added to define close relatives |
| 10/26/2022 | Annual review, no change to policy intent. Updating title, rationale, references and adding 81479 to coding. |
| 10/01/2021 |
Annual review, no change to policy intent. Updating policy number, background, raitonale and references. |
| 10/23/2020 |
Annual review, no change to policy intent, one criteria rewritten for clarity. Updating rationale, references and coding. |
| 10/15/2019 |
Annual review, no change to policy intent. Reformatting for clarity. |
| 10/29/2018 |
Annual review, no change to policy intent. |
| 11/02/2017 |
Annual review, rewriting policy for clarity, but, no changes to intent. |
| 04/26/2017 |
Updated category to Laboratory. No other changes. |
| 07/01/2016 |
NEW POLICY |