
Genetic Testing for Inherited Cardiomyopathies and Channelopathies - CAM 276
Description
Cardiomyopathies are diseases of the heart muscle. These conditions are frequently genetic and do not include muscle abnormalities caused by coronary artery disease, hypertension, valvular disease, and congenital heart disease. Symptoms include arrhythmia, cardiac dysfunction, and heart failure (Cooper Jr, 2022).
Channelopathies, also known as primary electrical disease, are a group of cardiac diseases caused by genetic defects in ion channels of the heart leading to arrhythmias, syncope, and the risk of sudden cardiac death (SCD) (Campuzano et al., 2015).
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request
- Genetic counseling IS REQUIRED for individuals prior to and after undergoing genetic testing for diagnostic, carrier, and/or risk assessment purposes.
- Sequencing of long QT syndrome (LQTS)-associated genes is considered MEDICALLY NECESSARY for the following:
- For individuals who have had a syncopal event and who have a Schwartz score greater than 1.
- For individuals for whom a cardiologist has a strong clinical suspicion of LQTS based on the individual’s clinical history, family history, and electrocardiogram (ECG) findings (e.g., Schwartz score greater than or equal to 3.5).
- For individuals with QT prolongation (QTc greater than 480 ms for prepubescent individuals, QTc greater than 500 ms for postpubescent individuals) in the absence of other clinical conditions that might prolong the QT interval (e.g., electrolyte abnormalities, hypertrophy, bundle branch block).
- For individuals with a close relative (see Note 1) with confirmed LQTS and for whom the familial mutation is not known.
- For close relatives (see Note 1) of an individual with a documented LQTS-causing mutation, testing for the known familial likely pathogenic or pathogenic variant is considered MEDICALLY NECESSARY.
- For individuals with a negative sequence analysis where the clinical suspicion of congenital LQTS remains high based on a Schwartz score greater than 1, genetic testing for LQTS with duplication/deletion analysis is considered MEDICALLY NECESSARY.
- Genetic testing for catecholaminergic polymorphic ventricular tachycardia (CPVT) is considered MEDICALLY NECESSARY in the following situations:
- For individuals who have a close relative (see Note 1) with a known CPVT likely pathogenic or pathogenic variant.
- For individuals who have a close relative (see Note 1) diagnosed with CPVT by clinical means but whose genetic status is unavailable.
- For individuals with signs and/or symptoms indicating a moderate-to-high pretest probability of CPVT, but for whom a definitive diagnosis cannot be made without genetic testing.
- For individuals who display exercise-, catecholamine-, or emotion-induced PVT or ventricular fibrillation in a structurally normal heart.
- Genetic testing for Brugada syndrome is considered MEDICALLY NECESSARY in the following situations:
- For individuals with signs and/or symptoms consistent with Brugada Syndrome but for whom a definitive diagnosis cannot be made without genetic testing.
- For individuals who have a close relative (see Note 1) with a known Brugada Syndrome likely pathogenic or pathogenic variant.
- Sequencing of short QT syndrome (SQTS)-associated genes is considered MEDICALLY NECESSARY for the following:
- For individuals for whom a cardiologist has a strong clinical suspicion of SQTS based on the individual’s clinical history, family history, and ECG findings (e.g., abnormally short QT intervals [less than or equal to 360 ms in males; less than or equal to 370 ms in females], an increased propensity to develop atrial and ventricular tachyarrhythmia in the absence of structural heart disease).
- Testing for the known familial likely pathogenic or pathogenic variant in asymptomatic individuals with a close relative (see Note 1) with a known SQTS likely pathogenic or pathogenic variant.
- For individuals with dilated cardiomyopathy and significant cardiac conduction disease (i.e., first-, second-, or third-degree heart block) and/or who have one or more family members who experienced sudden cardiac death or developed unexplained heart failure before 60 years of age, genetic testing for dilated cardiomyopathy is considered MEDICALLY NECESSARY.
- In asymptomatic close relatives (see Note 1) of an affected individual, genetic testing for a known familial likely pathogenic or pathogenic variant associated with dilated cardiomyopathy is considered MEDICALLY NECESSARY.
- Genetic testing for arrhythmogenic right ventricular cardiomyopathy (ARVC) is considered MEDICALLY NECESSARY in the following situations:
- For individuals with signs and/or symptoms consistent with ARVC but for whom a definitive diagnosis cannot be made without genetic testing.
- For individuals who have a close relative (see Note 1) with a known ARVC likely pathogenic or pathogenic variant.
- In asymptomatic close relatives (see Note 1) of an affected individual, genetic testing for a known familial likely pathogenic or pathogenic variant associated with progressive cardiac conduction disease (CCD or Lev-Lenegre disease) is considered MEDICALLY NECESSARY.
- In asymptomatic close relatives (see Note 1) of an affected individual, genetic testing for a known familial likely pathogenic or pathogenic variant associated with restrictive cardiomyopathy (RCM) is considered MEDICALLY NECESSARY.
- Genetic testing for left ventricular noncompaction (LVNC) is considered MEDICALLY NECESSARY in the following situations:
- For individuals with signs and/or symptoms consistent with LVNC but for whom a definitive diagnosis cannot be made without genetic testing.
- For individuals who have a close relative (see Note 1) with a known LVNC likely pathogenic or pathogenic variant.
- For individuals who meet the diagnostic criteria for hypertrophic cardiomyopathy (HCM), genetic testing for predisposition to HCM is considered MEDICALLY NECESSARY.
- For individuals who have a first-degree relative with established HCM, genetic testing for the known familial likely pathogenic or pathogenic variant is considered MEDICALLY NECESSARY.
- For individuals with a family history of HCM in which a first-degree relative with HCM has tested negative for likely pathogenic or pathogenic variants, genetic testing for a predisposition to HCM is considered NOT MEDICALLY NECESSARY.
- For symptomatic individuals with a Schwartz score less than or equal to 1, genetic testing for LQTS is considered NOT MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- For individuals with known LQTS, genetic testing for LQTS to determine prognosis and/or direct therapy is considered NOT MEDICALLY NECESSARY.
- For all other situations when the above criteria are not met, genetic testing for LQTS or CPVT is considered NOT MEDICALLY NECESSARY.
- For all other situations not meeting the criteria outlined above, genetic testing for Brugada Syndrome is considered NOT MEDICALLY NECESSARY.
- For all other situations not meeting the criteria outlined above, genetic testing for SQTS is considered NOT MEDICALLY NECESSARY.
- Genetic testing for Early Repolarization “J-wave” Syndrome, Sinus Node Dysfunction (SND), and/or other rhythm disorders is considered NOT MEDICALLY NECESSARY.
- For all other individuals, genetic testing for a predisposition to HCM is considered NOT MEDICALLY NECESSARY.
NOTES:
Note 1:Close blood relatives include first-degree relatives (e.g., parents, siblings, and children), second-degree relatives (e.g., grandparents, aunts, uncles, nieces, nephews, grandchildren, and half-siblings), and third-degree relatives (great-grandparents, great-aunts, great-uncles, great-grandchildren, and first cousins).
Table of Terminology
| Term |
Definition |
| ACC |
American College of Cardiology |
| ACM |
Arrhythmic cardiomyopathy |
| ACCF |
American College of Cardiology Foundation |
| ACMG |
American College of Medical Genetics and Genomics |
| ACTA2 |
Actin alpha 2, smooth muscle gene |
| ACTC1 |
Actin alpha cardiac muscle 1 gene |
| AF |
Atrial fibrillation |
| APHRS | Asia Pacific Heart Rhythm Society |
| APOB |
Apolipoprotein B gene |
| ARVC |
Arrhythmogenic right ventricular cardiomyopathy |
| ARVC/D |
Arrhythmogenic right ventricular cardiomyopathy/dysplasia |
| BrS |
Brugada syndrome |
| CASQ2 |
Calsequestrin 2 gene |
| CCD |
Cardiac conduction disease |
| CLQTS |
Congenital long QT syndrome |
| COL3A1 |
Collagen type III alpha 1 chain gene |
| CPVT |
Catecholaminergic polymorphic ventricular tachycardia |
| DCM |
Dilated cardiomyopathy |
| DSC2 |
Desmocoilin 2 gene |
| DSG2 |
Desmoglein 2 gene |
| DSP |
Desmoplakin gene |
| ECG |
Electrocardiogram |
| ECS |
European Society of Cardiology |
| EHRA |
European Heart Rhythm Association |
| ER | Early rpolarization |
| FBN1 |
Fibrillin-1 gene |
| FLNC |
Filamin C gene |
| HCM |
Hypertrophic cardiomyopathy |
| HF |
Heart failure |
| HFSA |
Heart Failure Society of America |
| HRS |
Heart Rhythm Society |
| IVF | Idiopathic ventricular fibrillation |
| KCNE1 |
Potassium voltage-gated channel subfamily E regulatory subunit 1 gene |
| KCNE2 |
Potassium voltage-gated channel subfamily E regulatory subunit 2 gene |
| KCNH2 |
Potassium voltage-gated channel subfamily H member 2 gene |
| KCNQ1 |
Potassium voltage-gated channel subfamily Q member 1 gene |
| LDLR |
Low density lipoprotein receptor gene |
| LMNA |
Lamin A/C gene |
| LQTS |
Long QT syndrome |
| LV |
Left ventricle |
| LVH |
Left ventricular hypertrophy |
| LVNC |
Left ventricular noncompaction |
| MYBPC3 |
Myosin binding protein C3 gene |
| MYH7 |
Myosin heavy chain 7 gene |
| MYH11 |
Myosin heavy chain 11 gene |
| MYL2 |
Myosin light chain 2 gene |
| MYL3 |
Myosin light chain 3 gene |
| PCCD | Progressive cardiac conduction disease |
| PCSK9 |
Proprotein convertase subtilisin/kexin type 9 gene |
| PKP2 |
Plakophilin 2 gene |
| PRKAG2 |
Protein kinase AMP-activated non-catalytic subunit gamma 2 gene |
| RCM |
Restrictive cardiomyopathy |
| RYR2 |
Ryanodine receptor 2 gene |
| SCA |
Sudden cardiac arrest |
| SCD |
Sudden cardiac death |
| SCN5A |
Sodium voltage-gated channel alpha subunit 5 gene |
| SMAD3 |
SMAD family member 3 gene |
| SND |
Sinus node dysfunction |
| SUDS | Sudden unexplaiined death syndrome |
| SQTS |
Short QT syndrome |
| TGFBR1 |
Transforming growth factor beta receptor 1 gene |
| TGFBR2 |
Transforming growth factor beta receptor 2 gene |
| TMEM43 |
Transmembrane protein 43 gene |
| TNNI3 |
Troponin I3, cardiac type gene |
| TNNT2 |
Troponin T2, cardiac type gene |
| TPM1 |
Tropomyosin 1 gene |
| TRDN |
Triadin gene |
| TTN |
Titin gene |
| WGS |
Whole genome sequencing |
Rationale
Cardiomyopathies
In 1995, the World Health Organization (WHO) divided cardiomyopathies into five categories: dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D), and unclassified cardiomyopathies. Cardiomyopathies have a variety of genotypes and phenotypes that typically require echocardiographic evaluation. Either phase of the heartbeat (systole or diastole) may be affected by cardiomyopathies (Cooper Jr, 2022).
Systolic dysfunction is usually characterized by a decrease in myocardial contractility. This decrease causes a reduction in the ejection fraction of the left ventricle (LV), thereby forcing one of two compensatory mechanisms; either the LV itself increases in size (leading to larger stroke volume) or the contractility of the heart increases in response to increased stretch. However, these compensatory mechanisms will eventually fail, leading to physiological manifestations of heart failure. This dysfunction is often seen in DCM, as well as some HCM patients (Cooper Jr, 2022).
Diastolic dysfunction refers to abnormal LV relaxation and filling, as well as elevated filling pressures. As with systolic dysfunction, the primary issue with diastole is caused by abnormal contractility of the heart muscle. The contractility of the myocardium influences both the LV relaxation phase (the isovolumetric period between the aortic valve’s close and mitral valve’s opening) and passive compliance phase (variable pressure, starting at mitral valve’s opening) of diastole. Due to the impaired contractility of the myocardium, the pressure in each phase is abnormal. Dysfunction in this phase is often seen in HCM, RCM, and DCM (Cooper Jr, 2022).
Hypertrophic cardiomyopathy (HCM)
Hypertrophic cardiomyopathy (HCM) is a commonly inherited cardiovascular disease defined as thickening of the ventricular wall resulting from more than 1500 mutations in 11 or more genes encoding proteins of the cardiac sarcomere (Maron, 2022).
Hypertrophic cardiomyopathy (HCM) is characterized by left ventricular hypertrophy (LVH, thickness of ≥ 15 mm), observed by echocardiography or magnetic resonance imaging and not otherwise explainable by other cardiovascular issues, such as coronary artery disease, hypertension, valvular disease, and congenital heart disease. Development of LVH usually starts in adolescence and is complete by early adulthood. Symptoms include chest pain, dyspnea and syncope, and severe disease can lead to disabling complications, including heart failure and malignant ventricular arrhythmias. However, many patients with HCM are asymptomatic or have minimal symptoms and are only discovered through means such as family screenings or an abnormal ECG (Maron, 2022). HCM is the most frequent cause of sudden death in young people and can lead to functional disability from heart failure and stroke (Maron, 2003). HCM has a prevalence of approximately 1 in 500 people (Maron et al., 1995). However, estimates of clinically expressed HCM plus gene carriers are as high as 1 in 200 (Semsarian et al., 2015).
More than 90% of HCM is inherited as an autosomal‐dominant disease with variable expressivity and age‐related penetrance (Frustaci et al., 2018). Currently, relevant genetic abnormalities can be detected in approximately 60 percent of patients with clinically documented HCM (Cirino et al., 2017; Maron et al., 2012). Most of the genetic mutations associated with HCM are found in the genes encoding various proteins that make up the cardiac sarcomere, the basic contractile unit of cardiac myocytes. More than 1500 pathogenic variants have been identified in at least 11 different genes (Cirino, 2014; Maron et al., 2012). Mutations in myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) are the most common and account for roughly 70 percent of HCM. Other genes implicated in HCM are regulatory myosin light chain (MYL2) and cardiac troponin T (TNNT2). Non-sarcomeric genes encoding plasma membrane or mitochondrial proteins, or Z‐disc encoding genes, have also been documented (Frustaci et al., 2018).
Wide phenotypic variability exists, ranging from asymptomatic to severe life-threatening heart failure even within the same mutation. This variability in clinical expression may be related to environmental factors and modifier genes (Alcalai et al., 2008). Moreover, no strong correlation between left ventricular problems and symptoms exists; patients with major obstructions or hypertrophy may be asymptomatic and vice versa. The primary characteristic of LVH is present in multiple conditions, such as systemic hypertension, Fabry disease, aortic stenosis, and more. Such conditions should be excluded before a diagnosis of HCM is made (Maron, 2022).
Diagnostic screening of first-degree relatives is important to identify at-risk patients. Guidelines have been established for clinically unaffected relatives of affected individuals. Clinical screening with physical examination, electrocardiography, and echocardiography is recommended every 12 to 18 months for individuals between the ages of 12 to 18 years and every three to five years for adults with additional screening recommended for any change in symptoms (Gersh et al., 2011).
Dilated cardiomyopathy (DCM)
Dilated cardiomyopathy (DCM) is characterized by dilation and impaired contraction of one or both ventricles. The dilation often becomes severe and is invariably accompanied by an increase in total cardiac mass. Affected patients have impaired systolic function and clinical presentation is usually with features of heart failure (Cooper Jr., 2022).
Dilated cardiomyopathy is caused by a variety of disorders. The cause is unknown for over 50% of patients with the disease. Familial dilated cardiomyopathy is caused by a genetic mutation in 20-35% with the disease. DCM is transmitted primarily in an autosomal dominant inheritance pattern. Mutations in over 30 genes have been determined to cause familial dilated cardiomyopathy (Hershberger, 2023).
Other common causes for DCM include (AHA, 2024) coronary heart disease, heart attack, high blood pressure, diabetes, thyroid disease, viral hepatitis and HIV, infections, especially viral infections that inflame the heart muscle, alcohol, complications during the last month of pregnancy or within five months of birth, certain toxins such as cobalt, certain drugs (such as cocaine and amphetamines) and two medicines used to treat cancer (doxorubicin and daunorubicin).
Genetic forms of dilated cardiomyopathy are diagnosed by family history and molecular testing. Genes associated with familial dilated cardiomyopathy include MYH7, MYBPC3, TNNT2, TNNC1, TNNI3, TPM1, MYL2, MYL3, ACTC1, ACTN2, CSRP3, PLN, TTR, PRKG2, LAMP2, GLA, LMNA, BAG3, RBM20, SCN5A, DES, DSC2, DSG2, DSP, JUP, PKP2, RYR2, TMEM43, and TTN (Hershberger et al., 2018).
The frequencies of DCM mutations in any one gene are low (< 1% to 6% – 8%), and a genetic cause is identified in only 30-35% of familial DCM cases. Therefore, routine genetic testing for DCM was only recommended in familial disease (≥ 2 affected family members) (Yancy et al., 2013). However, as molecular genetic testing laboratories offer DCM genetic testing panels of 12 – 30 genes utilizing next-generation sequencing methods, testing sensitivity now ranges from 15% – 25%, has become standard of care (M. J. Ackerman et al., 2011; Hershberger, 2023; Hershberger et al., 2009; Hershberger et al., 2010; Hershberger & Siegfried, 2011).
Guidelines now recommend testing for all patients with cardiomyopathy even if no other disease is evident in the family. Genetic testing is clinically useful in the management of affected individuals, as well as to assess risk in relatives (Hershberger et al., 2018). LMNA mutations are associated with high rates of conduction system disease, ventricular arrhythmias, and sudden cardiac death (SCD), and may consequently lower the threshold for prophylactic ICD implantation. DCM patients with a variant of the SCN5A gene exhibit a phenotype associated with significant arrhythmias and frequent premature ventricular complexes. Although such patients responded poorly to conventional HF therapy, treatment with sodium channel blocking drugs produced a dramatic reduction in ectopy and normalization of left ventricular (LV) function (Japp et al., 2016).
Restrictive cardiomyopathy (RCM)
Restrictive cardiomyopathy (RCM) differs from other cardiomyopathies in that there may not be many physical abnormalities (i.e., no dilation or hypertrophy). However, the ventricular filling process is still significantly impaired. RCM may be difficult to see on two-dimensional imaging, and assessment of flow velocity across the mitral valve is more accurate in detecting these filling abnormalities (Cooper Jr, 2022). Some cases of RCM are secondary to a more apparent cardiac condition. However, idiopathic cases of RCM are generally genetic conditions. Pathogenic mutations in sarcomeric or cytoskeletal genes such as TNNI3, TNNT2, and TPN1 have been linked to familial RCM (Ammash, 2024).
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D)
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is characterized by changes in myocardium of the right (and often left as well) ventricular free wall. This myocardium is replaced by fibrous or fibro-fatty tissue, causing dysfunction of the affected ventricles (Cooper Jr., 2022). At the molecular level, the desmosomes are typically impaired due to genetic mutations. This causes the mechanical stress of the heart to detach myocytes, which eventually leads to their death. The initial repair mechanisms of this detachment are what produces the fibrous tissue. Up to 30% of ARVC cases are familial, and mutations in gene products such as plakoglobin and desmoplakin have been associated with ARVC (McKenna, 2024).
Unclassified Cardiomyopathies
Other types of cardiomyopathies that do not fall into one of the other four categories are considered “unclassified.” For example, left ventricular noncompaction (LVNC) falls in this category due to its characteristic myocardial wall; the myocardial wall consists of “prominent trabeculae and deep intertrabecular recesses,” thereby resulting in two layers of myocardium of different thickness. This condition may be sporadic or familial; up to 50% of LVNC cases are familial. Genes such as alpha-dystrobrevin or other sarcomeric genes may contribute to LVNC (Attenhofer-Jost & Connolly, 2022).
This category also includes cardiomyopathies such as stress-induced cardiomyopathy and cirrhotic cardiomyopathy (Cooper Jr, 2022).
Clinical Utility and Validity (Cardiomyopathies)
A study by Cirino et al. (2017) compared the results from panel genetic testing to whole genome sequencing (WGS). Forty-one patients with HCM who had undergone targeted genetic testing (either multigene panel or familial variant test) were recruited into a clinical trial of WGS. Panel size ranged from 4 – 62 genes, and all but two subjects were tested for the main eight sarcomeric genes (MYH7, MYBPC3, TNNT2, TPM1, MYL2, MYL3, TNNI3, ACTC). The authors stated that WGS detected nearly all variants identified on panel testing and allowed further analysis of posited disease genes. Several variants of uncertain clinical use and other genetic findings were also identified. Panel testing and WGS provided similar results, but WGS provides reanalysis over time; however, WGS also requires genomic expertise to correctly interpret results (Cirino et al., 2017).
A study focusing on the non-sarcomeric genes contributing to HCM was performed by Walsh et al. (2017). A reference sample of 60,706 exomes were analyzed and compared to 6,179 HCM cases. This comparison revealed many gene variants in the main eight sarcomeric genes (MYH7, MYBPC3, TNNT2, TPM1, MYL2, MYL3, TNNI3, ACTC1) but very few variants of the non-sarcomeric genes in HCM cases. The authors concluded the variation in most of the non-sarcomeric genes does not affect HCM significantly as 99% of HCM pathogenic variants were found to be in the main eight sarcomeric genes. Four non-sarcomeric genes were found to have an excess of variants, but even these amounted to only 2% of the HCM cases overall; the other 26 non-sarcomeric genes examined were found to have very little or no excess variation over the reference sample of exomes. Furthermore, the authors state that only the well-known variants are symptomatic whereas the other variants are of unknown significance or benign, making clinical sequencing of limited use. The authors recommended that the only genes tested should be the eight sarcomeric genes, the metabolic cardiomyopathy genes, and possibly ACTN2 and MYOZ2 (Walsh et al., 2017).
Bhonsale et al. (2015) assessed the impact of genotype on clinical outcomes of ARVC patients. Pathogenic mutations were evaluated in 577 patients. The investigators found that patients with a desmoplakin-associated mutation had an over four-fold occurrence of left ventricular (LV) dysfunction and heart failure than PKP2 carriers. No significant difference was found between clinical outcomes of patients with missense mutations and patients with truncating or splice site mutations. Patients with multiple mutations had more severe symptoms, such as lower survival rate without ventricular fibrillation or tachycardia, more frequent LV dysfunction, heart failure, cardiac transplant, and earlier occurrence of sustained ventricular fibrillation and tachycardia, as compared to those with only one mutation (Bhonsale et al., 2015).
Kostareva et al. (2016) evaluated the “genetic spectrum” of idiopathic RCM. The authors screened for 108 cardiomyopathy and arrhythmia-associated genes in 24 patients with idiopathic RCM. They found pathogenic and “likely-pathogenic” variants in 13 of the 24 patients (54%), and half of these genotype-positive patients carried a combination of pathogenic variants, likely-pathogenic variants, and variants of unknown significance. The most frequent combination included mutations in sarcomeric and cytoskeletal genes (Kostareva et al., 2016).
Kayvanpour et al. (2017) evaluated the genetic-phenotype associations for DCM. The study included 48 studies encompassing 8097 patients, and the authors investigated mutations in LMNA, PLN, RBM20, MYBPC3, MYH7, TNNT2 and TNNI3. The authors results were as follows: “The average frequency of mutations in the investigated genes was between 1 and 5 %. The mean age of DCM onset was the beginning of the fifth decade for all genes. Heart transplantation (HTx) rate was highest in LMNA mutation carriers (27 %), while RBM20 mutation carriers were transplanted at a markedly younger age (mean 28.5 years). While 73 % of DCM patients with LMNA mutations showed cardiac conduction diseases, low voltage was the reported ECG hallmark in PLN mutation carriers. The frequency of ventricular arrhythmia in DCM patients with LMNA (50 %) and PLN (43 %) mutations was significantly higher. The penetrance of DCM phenotype in subjects with TTN truncating variants increased with age and reached 100 % by age of 70” (Kayvanpour et al., 2017).
Genetic testing has been recognized as a valuable part of diagnosis and classification in pediatric cardiomyopathy. Lipshultz et al. (2019) identified a single-center study of 63 children with cardiomyopathy that reported that 42% of the illness could be attributed to a genetic cause, as defined by the presence of an affected first-degree family member having tested positive for an HCM or DCM gene panel mutation. However, it was noted that though the morphofunctional phenotype in children may be similar to those of adults, the causes and proportion resulting from them differ. These differences are believed to be neuromuscular, metabolic, mitochondrial, and syndromic causes also playing important roles in children. Lipshultz et al. (2019) identified another study to corroborate this finding: in 916 children with an identified case of cardiomyopathy, an underlying metabolic or syndromic cause was present in more than a third, and a metabolic or syndromic cause was present in 40% to 50% of 61 children with HCM and 8% of 1731 children with DCM in the Pediatric Cardiomyopathy Registry (Lipshultz Steven et al., 2019).
Parker and Landstrom (2021) reviewed current literature on what role genetic testing play in diagnosing and managing patients and families with a history of pediatric CM. With relation to HCM, the researchers identified multiple studies demonstrating that patients with “sarcomere variants tend to be more severely affected than patients with non-sarcomeric variants” and “within the sarcomere genes, some argue that specific sarcomere variants are inherently more pathogenic than others.” On top of that, there appear to be some other genetic and environmental factors that influence the HCM phenotype in each patient, thus indicating that the genotype-phenotype correlation could greatly affect the clinical course and prognosis. Especially for metabolic forms of HCM and patients with a clear genetic etiology, treatment may be adopted based on these genetic findings; for instance, “in GAA-positive Pompe disease, alpha-glucosidase enzyme replacement therapy is associated with HCM reversal in affected children” (Parker & Landstrom, 2021).
Similar concepts were mentioned for DCM, as some genotypes yielded worse phenotypes. For example, “TNNC1 variants are associated with early onset DCM, and increased rates of heart transplant and SCD compared to other genes.” Gene therapy could also be explored as an option, such as for Duchenne muscular dystrophy, to “target and rescue functional expression of pathogenic variants.” For arrhythmic cardiomyopathy (ACM), Parker and Landstrom (2021) specifically mention how confirming an ACM diagnosis via genetic testing “versus an arrhythmic syndrome secondary to a cardiac channelopathy” could aid clinical management. Variants in the PKP2 and TMEM43 genes have been found to be associated with exercise-induced disease progression and risk of sudden cardiac arrest (SCA), so this could impact chosen involvement in sports (Parker & Landstrom, 2021).
Grondin et al. (2022) studied the clinical utility of genetic testing in unexplained cardiac arrest. The study included 228 unexplained cardiac arrest survivors. Whole exome sequencing was performed on each participant to identify pathogenic or likely-pathogenic variants. In total, 23 of the 228 participants (10%) had a pathogenic or likely-pathogenic variant. Genetic testing increased the proportion of “explained” cases from 9%, with only phenotyping, to 18%, with phenotyping and WES. “The majority of disease-causing variants was located in cardiomyopathy-associated genes, highlighting the arrhythmogenic potential of such variants in the absence of an overt cardiomyopathy diagnosis” (Grondin et al., 2022).
Cardiac Ion Channelopathies
The electromechanical pumping action of the heart maintains circulation and ensures the delivery of blood and nutrients to all organs to support their normal function. Synchronized contraction of the myocardium is necessary to generate sufficient pressure to drive blood flow (Voorhees & Han, 2015). Mechanical contraction of cardiac myocytes is coordinated by the generation and propagation of an action potential (Fernandez-Falgueras et al., 2017) through the synergistic activation and inactivation of several voltage-dependent ion channels. Membrane depolarization during the action potential leads to the opening of the voltage-gated calcium channels resulting in an inward current, followed by the efflux of potassium ions, generation of an outward current, and cell repolarization (Garcia-Elias & Benito, 2018). Action potential duration is determined by the magnitude and timing of inward and outward currents (Kirk & Kass, 2015). Differential expression, selectivity, and gating properties of cardiac ion channels in distinct regions of the heart promote unidirectional propagation of electrical activity (Fernandez-Falgueras et al., 2017).
Mutations in genes encoding these specific channels or associated proteins may impair ionic conduction resulting in changes in action potential, synchronization, and/or propagation of electrical impulse and predispose to potentially malignant arrhythmias (Nerbonne & Kass, 2005; Roden et al., 2002). Dyssynchronous contraction of the ventricle, arising from electrical activation delays, also significantly worsens morbidity and mortality in heart failure (HF) patients (Kirk & Kass, 2015). Ion channelopathies have been identified as a significant cause of sudden cardiac death (SCD) in patients with structurally normal hearts (Campuzano et al., 2015; Magi et al., 2017), and some cases of otherwise unexplained stillbirth (Munroe et al., 2018).
Patients can show early symptoms of palpitations or hemodynamic compromise, including dizziness, seizure, or syncope, particularly following exertion; however, in many cases SCD is the only sign of cardiac trouble (Martin et al., 2013). Electrical disturbances in the heart rhythm that can be detected on electrocardiogram (ECG) of some patients with channelopathies result in diagnosis of:
- Long QT Syndrome (LQTS) is characterized by prolonged ventricular repolarization and electrocardiographic prolongation of the QT interval (QTc ≥ 480 ms in repeated 12-lead ECG, although a QTc ≥ 460 ms is sufficient in the presence of unexplained syncope). The variable clinical manifestations of LQTS range from asymptomatic patients diagnosed through family screening, to SCD, syncope, convulsions, malignant ventricular arrhythmias, VF, and torsade de pointes (Fernandez-Falgueras et al., 2017). The prevalence of LQTS in infants is approximately 1 in 2000 (Schwartz et al., 2009).
- Diagnosis of LQTS is made using the Schwartz Score, which indicates high probability of LQTS with a score ≥3.5. The table of diagnostic criteria is shown below (Schwartz & Crotti, 2011)

In the absence of medications or disorders known to affect these electrocardiographic features.
^ QTc calculated by Bazett’s formula where QTc = QT/√RR.
* Mutually exclusive.
Resting heart rate below the 2nd percentile for age.
The same family member cannot be counted in A and B.
SCORE: ≤ 1 point: low probability of LQTS.
1.5 to 3 points: intermediate probability of LQTS.
>3.5 points high probability.
- Brugada Syndrome (BrS) is clinically characterized by right ventricular conduction delay and ST-segment elevation in the anterior right precordial leads. Syncope is one of the main clinical manifestations; individuals with BrS develop a monomorphic ventricular tachycardia that may precipitate during sleep, rest, or fever (Magi et al., 2017). Recent reports suggest that BrS could be responsible for 4%–12% of all sudden death (SD) and up to 20% of SD in patients with structurally normal hearts (Fernandez-Falgueras et al., 2017).
- Short QT Syndrome (SQTS) is characterized by abnormally short QT intervals and an increased propensity to develop atrial and ventricular tachyarrhythmia in the absence of structural heart disease. Cardiac arrest seems to be the most frequent symptom (up to 40%). Palpitations are a common symptom (30%), followed by syncope (25%) and atrial fibrillation (AF), which are the first symptoms of the disease in up to 20% of patients. The episodes may occur in a wide range of situations, such as in reaction to loud noise, at rest, during exercise, and during daily activity (Fernandez-Falgueras et al., 2017)
- Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) is characterized by a normal ECG and ventricular arrhythmia in genetically predisposed individuals during intense physical exercise or acute emotional stress. Typical clinical manifestations of CPVT include dizziness and syncope. Ventricular arrhythmia, however, may degenerate into rapid polymorphic ventricular tachycardia and ventricular fibrillation, leading to SCD (Magi et al., 2017).
- Progressive Cardiac Conduction Disease (PCCD) is characterized by problems with the cardiac impulse of the heart. These conduction abnormalities may be accompanied by structural problems such as fibrous or fatty calcification, and an ECG may display unusual patterns such as a prolonged P-wave or prolonged QRS interval. These arrhythmias may lead to sudden cardiac death. Mutations in ion channel proteins such as TRPM4 or SCN5A have been associated with CCDs (M. J. Ackerman et al., 2011; Wilde et al., 2005).
Not all cases are accompanied by changes in ECG, which makes them more difficult to diagnose. Genetic testing can contribute substantially both to the diagnosis of affected patients and with the identification of asymptomatic individuals at risk (Bastiaenen & Behr, 2011; Priori et al., 2013).
Currently, mutations associated with SCD have been identified in sodium, potassium and calcium channels and associated proteins (Fernandez-Falgueras et al., 2017). A general overview of the main genetic variants that have been linked to the major cardiac channelopathies is displayed in the table below [adapted from (Garcia-Elias & Benito, 2018; Magi et al., 2017; Munroe et al., 2018; Tester & Ackerman, 2011)].
| Gene |
Protein |
Prevalence |
Other Associations |
| Brugada Syndrome (BrS) |
|||
|
|
Ion Channel Subunits |
|
|
| SCN5A* |
NaV1.5 (α-subunit of the voltage-dependent Na+channel) |
⋍ 25% (BrS1) |
DCM, ARVC, Heart block, LQTS, SSS, SIDS |
| SCN1B* |
β1-subunit of the voltage-dependent Na+ channel |
< 1% |
CCD, Epilepsy |
| SCN2B* |
β2-subunit of the voltage-dependent Na+ channel |
< 1% |
AF |
| SCN3B* |
β3-subunit of the voltage-dependent Na+ channel |
< 1% |
AF, VF, SIDS |
| SCN10A* |
NaV1.8 (α-subunit of the neuronal voltage-dependent Na+ channel) |
⋍ 10% |
LQTS, AF, painful small-fiber peripheral neuropathy |
| CACNA1C* |
CaV1.2 (α1C-subunit of the volatge-dependent L-type Ca2+ channel) |
< 1% |
Timothy syndrome, LQTS |
| CACNB2b* |
β2-subunit of the voltage-dependent L-type Ca2+channel |
< 1% |
SQTS |
| KCND3* |
KV4.3 (α-subunit of the voltage-dependent K+channel) |
< 1% |
SIDS, Spinocerebellar ataxia |
| KCNE3* |
minK-related peptide 2 (β-subunit of the voltage-dependent K+ channel) |
< 1% |
|
| KCNAB2 |
β2-subunit of the voltage-dependent K+ channel |
< 1% |
|
| KCND2 |
KV4.2 (voltage-dependent K+ channel) |
< 1% |
Epilepsy |
| KCNE5* |
minK-related peptide 4 (β-subunit of the voltage-dependent K+ channel) |
< 1% |
AF, VF |
| KCNJ8* |
Kir6.1 (inward-rectifier K+channel, subunit of the ATP-sensitive K+ channel) |
< 1% |
VF, SIDS, Cantu syndrome |
| ABCC9* |
SUR2 (sulfonylurea receptor, subunit of the ATP-sensitive K+ channel) |
< 1% |
DCM, ERS, Cantu syndrome and |
| KCNH2* |
KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) |
< 1% |
LQTS, SQTS |
| CACNA2D1* |
α2/δ subunit of the volatge-dependent L-type Ca2+channel |
< 1% |
Epilepsy |
| HCN4* |
hyperpolarization-activated, cyclic nucleotide-gated ion channel 4 |
< 1% |
SSS, AF, AV block, Bradycardia, |
| TRPM4* |
Transient receptor potential melastatin 4 |
< 1% |
Herat Block, LQTS |
|
|
Auxiliary Proteins |
|
|
| FGF12 |
fibroblast growth factor 12 |
< 1% |
Epilepsy |
| GPD1L* |
glycerol-3-phosphate dehydrogenase 1-like |
< 1% |
|
| SLMAP |
sarcolemma associated protein (striatin-interacting phosphatase and kinase complex) |
< 1% |
|
| PKP2* |
plakophillin-2 |
< 1% |
ARVC |
| SEMA3A |
semaphorin-3A |
< 1% |
|
| RANGRF* |
MOG1 (multicopy suppressor of Gsp1) |
< 1% |
histiocytoid cardiomyopathy |
| HEY2 |
CHF1 (cardiovascular helix-loop-helix factor 1) |
< 1% |
|
| Long QT Syndrome (LQTS) |
|||
|
|
Ion Channel Subunits |
|
|
| KCNQ1* |
KV7.1 (α-subunit of the voltage-dependent K+ channel) |
⋍ 40% (LQT1) |
JLNS, SQTS |
| KCNH2* |
KV11.1/hERG (α-subunit of the voltage-dependent K+channel) |
⋍ 30% (LQT2) |
SQTS |
| SCN5A* |
NaV1.5 (α-subunit of the voltage-dependent Na+ channel) |
⋍ 10% (LQT3) |
BrS, DCM, ARVC, Heart block, SSS, SIDS |
| KCNE1* |
minK (β1-subunit of the voltage-dependent K+ channel) |
< 1% |
JLNS |
| KCNE2* |
MiRP1 (β2-subunit of the voltage-dependent K+ channel) |
< 1% |
|
| KCNJ2* |
Kir2.1 (inward rectifying K+ channel) |
< 1% (LQT7) |
Andersen-Tawil syndrome, SQTS, AF |
| KCNJ5* |
Kir3.4 (G protein-activated inward rectifying K+ channel 4) |
< 1% |
LQTS, Hyperaldosteronism |
| SCN1B* |
β1-subunit of the voltage-dependent Na+ channel |
< 1% |
BrS, CCD, Epilepsy |
| SCN4B* |
β4-subunit of the voltage-dependent Na+ channel |
< 1% |
AF |
| CACNA1C* |
CaV1.2 (α1C-subunit of the voltage-dependent L-type Ca2+ channel) |
< 1% (LQT8) |
BrS, Timothy syndrome |
|
|
Auxiliary Proteins |
|
|
| AKAP9* |
A-kinase anchor protein-9 |
< 1% |
|
| ANK2* |
ankyrin B |
< 1% |
Arrhythmia |
| CALM1* |
calmodulin (CaM) |
< 1% |
CVPT |
| CALM2* |
calmodulin (CaM) |
< 1% |
CVPT |
| CALM3* |
calmodulin (CaM) |
< 1% |
CVPT |
| SNTA1* |
α1-syntrophin |
< 1% |
|
| TRDN* |
triadin |
< 1% |
CVPT |
| CAV3* |
caveolin-3 |
< 1% |
HCM, LGMD, Rippling muscle disease, Tateyama-type distal myopathy, SIDS |
| TRPM4* |
Transient receptor potential melastatin 4 |
< 1% |
Herat Block, BrS |
| RYR2* |
ryanodine receptor 2 (RyR2) |
< 1% |
ARVC, CPVT |
| Short QT Syndrome (SQTS) |
|||
|
|
Ion Channel Subunits |
|
|
| KCNH2* |
KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) |
⋍ 15% (SQT1) |
LQTS |
| KCNQ1* |
KV7.1 (α-subunit of the voltage-dependent K+channel) |
< 1% |
JLNS, LQTS |
| KCNJ2* |
Kir2.1 (inward rectifying K+ channel) |
< 1% |
Andersen-Tawil syndrome, AF |
| CACNA1C* |
CaV1.2 (α1C-subunit of the voltage-dependent L-type Ca2+ channel) |
< 1% |
BrS, Timothy syndrome |
| CACNB2b* |
β2-subunit of the voltage-dependent L-type Ca2+channel |
< 1% |
BrS |
| CACNA2D1* |
α2/δ-subunit of the voltage-dependent L-type Ca2+ channel |
< 1% |
BrS, Epilepsy |
| Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) |
|||
| RYR2* |
ryanodine receptor 2 (RyR2) |
⋍ 50% – 60% (CPVT1) |
ARVC |
| KCNJ2 |
Kir2.1 |
10% |
|
| CASQ2* |
calsequestrin 2 |
⋍ 5% |
|
| TRDN* |
triadin |
< 1% |
LQTS |
| CALM1* |
calmodulin (CaM) |
< 1% |
LQTS |
| CALM2* |
calmodulin (CaM) |
< 1% |
LQTS |
| CALM3* |
calmodulin (CaM) |
< 1% |
LQTS |
| TECLR |
trans-2,3-enoyl-CoA reductase- like |
< 1% |
|
| * *- commercially available test |
|||
| Abbreviations: AF – Atrial fibrillation; ARVC- Arrhythmogenic right ventricular cardiomyopathy; AV – Atrioventricular; BrS – Brugada syndrome; CCD – Cardiac conduction defect; CHD – Congenital heart defects; CPVT – Catecholaminergic polymorphic ventricular tachycardia; DCM – Dilated cardiomyopathy; EMD – Emery Dreifuss muscular dystrophy; ERS –Early repolarization syndrome; HCM – Hypertrophic cardiomyopathy; HCC - histiocytoid cardiomyopathy; JLNS – Jervell and Lange-Nielsen syndrome; LGMD – Limb girdle muscular dystrophy; LQTS – Long QT syndrome; SIDS – Sudden infant death syndrome; SQTS – Short QT syndrome; SSS – Sick sinus syndrome; SUDS – Sudden unexpected death syndrome; VF – Ventricular fibrillation |
|||
The clinical presentations of these conditions overlap as shown below (adapted from Campuzano, 2015), and genetic testing may clarify diagnoses, etiologies, and treatments in symptomatic individuals (Spoonamore & Johnson, 2016). However, predicting clinical presentation based on genetic mutation is also challenging (Bezzina et al., 2015) due to the incomplete penetrance of most of these genes (Giudicessi & Ackerman, 2013).
Gene Mutations and their Associated Conditions
| ABCC9 |
KCND3 |
SCN2B |
||
| FCF12 |
KCNE3 |
SCN3B |
||
| GPD1L |
KCNE5 |
SCN10A |
||
| BrS |
HCN4 |
KCNJ8 |
SEMA3A |
|
| HEY2 |
PKP2 |
SLAMP |
||
| KCND2 |
RANGRF |
TRPM4 |
||
| SCN1B |
CACNA1C |
CACNA2D1 |
||
| SCN5A |
CACNB2 |
|||
| ANK2 |
KCNQ1 |
|
SQTS |
|
|
|
AKAP9 |
KCNH2 |
|
|
| CAV3 |
KCNJ2 |
|
||
| KCNE1 |
RYR2 |
TRDN |
||
| LQTS |
KCNE2 |
CALM1 |
CASQ2 |
|
| KCNJ5 |
|
CPVT |
||
| SCN4B |
|
|||
| SNTA1 |
|
|||
| CALM2 |
|
Proprietary Testing (Channelopathies)
Proprietary gene panels exist for the assessment of cardiac ion channelopathies. For example, GeneDX offers several customizable panels for various channelopathies and cardiomyopathies. Conditions such as ARVC, Brugada Syndrome, and CPVT are available as separate panels, and GeneDX offers combined panels such as a “Custom Arrhythmia Panel”, a “Custom Cardiomyopathy Panel”, and a “Combined Cardiac Panel” (GeneDX, 2024). Other commercially available panels include offerings from Invitae (Invitae, 2024), Fulgent, (Fulgent, 2024) and BluePrint Genetics (BluePrint, 2024).
Analytical Validity (Channelopathies)
Ware et al. (2013) compared two NGS approaches for diagnostic sequencing inherited arrhythmia syndromes. PCR-based target enrichment and long-read sequencing (PCR-LR) was compared to in-solution hybridization-based enrichment and short-read sequencing (Hyb-SR). The PCR-LR assay comprehensively assessed five long QT genes (KCNQ1, KCNH2, SCN5A, KCNE1 and KCNE2) and "hot spots" in RYR2. The Hyb-SR assay targeted 49 genes, including those in the PCR-LR assay. The sensitivity for detection of control variants was identical. In both assays, the major limitation was upstream target capture, particularly in regions of extreme GC content. These initial experiences with NGS cardiovascular diagnostics achieved up to 89% sensitivity at a fraction of current costs (Ware et al., 2013).
Proost et al. (2017) validated a targeted gene panel for next-generation sequencing of 51 genes associated with primary electrical disease with 20 Human Polymorphism Study Center samples and 19 positive control samples with a total of 1479 variants. “An analytical sensitivity and specificity of 100% and 99.9% were obtained.” After validation, the assay was applied to “114 PED patients which identified 107 variants in 36 different genes, 18 of which were classified as pathogenic or likely pathogenic, 54 variants were of unknown significance, and 35 were classified as likely benign.” They concluded “that the PED Multiplex Amplification of Specific Targets for Resequencing Plus assay is a proficient and highly reliable test to routinely screen patients experiencing primary arrhythmias” (Proost et al., 2017).
Clinical Utility and Validity (Channelopathies)
Garcia et al. (2016) proposed a framework for establishing clinical validity for assessing polymorphisms of inherited cardiac conditions, as well as evaluating the strength of association between genotype and phenotype, from a logical argument. Clinical validity of a gene is established when a gene is known to cause disease; since a specific variant must be responsible for causing the disease, a gene variant must be known to cause that disease. Conversely, if no variants can be established to cause disease, the clinical validity association has not been established. Variants of unknown significance (VUS) would, therefore, not have established clinical validity. Garcia et al. (2016) proposed three categories of strength of association: strong, suggested, and emerging.
- “Strong” refers to “cases where there exists at least one clinically observed variant supported by sufficient evidence to classify that variant as pathogenic. “Strong” indicates that the relationship has been proven.”
- “Suggested” is used in cases where some preliminary evidence exists suggesting a causal relationship, but the relationship has not yet been formally proven.”
- “Emerging” is used to describe a growing suspicion that a specific condition is caused by a gene that has already been proven to cause disease.
The authors go on to state that if one “strong” relationship exists, then clinical validity has been established. If not, the gene should be considered of “uncertain significance” (Garcia et al., 2016).
The authors also provide recommendations for gene panels to test for certain conditions. They “propose that a comprehensive panel test designed for the molecular diagnosis of a particular condition should include the following classes of genes:
- Genes that have been conclusively proven to cause the condition in question.
- Genes suspected but not yet proven to cause the condition in question.
- Genes that have been conclusively proven to cause a condition within the clinical differential. This category should include genes that cause a condition that can progress into the condition in question, genes that cause a condition that can be confused with the condition in question, and genes that cause a syndrome that include the condition in question as a primary feature.
They suggest that the clinical validity of a panel is established when that panel includes a set of genes that account for a substantial proportion of the genetic causes of the disease in question. Conversely, a panel is NOT valid if it omits certain genes that account for a substantial proportion of the known genetic risk. A clinically valid panel may also include genes for which some preliminary evidence of clinical validity exists (“preliminary evidence genes”) (Garcia et al., 2016).
Hofman et al. (2013) analyzed the yield of DNA testing over 15 years. They analyzed results from 7021 individuals who were counseled, 6944 from 2298 different families (aged 41±19 years; 49% male). In 702 families (31%), a possible disease-causing mutation was detected. The yield of DNA testing of probands with primary electric diseases was 47% in LQTS, 26% in BrS, and 37% in CPVT. Cascade screening revealed 1539 mutation-positive subjects, and in 372 families counseled after sudden unexplained death an inherited arrhythmia syndrome was diagnosed in 29% (n=108) (Hofman et al., 2013).
Le Scouarnec et al. (2015) et al. examined 167 index cases presenting with a Brugada pattern on the electrocardiogram as well as 167 individuals aged over 65-years old and showing no history of cardiac arrhythmia. They found that “a significant enrichment in rare coding variation (with a minor allele frequency below 0.1%) was observed only for SCN5A, with rare coding variants carried by 20.4% of cases with BrS versus 2.4% of control individuals. No significant enrichment was observed for any other arrhythmia-susceptibility gene, including SCN10A and CACNA1C.
These results indicate that, except for SCN5A, rare coding variation in previously reported arrhythmia-susceptibility genes do not contribute significantly to the occurrence of BrS in a population with European ancestry. Extreme caution should thus be taken when interpreting genetic variation in molecular diagnostic setting, since rare coding variants were observed in a similar extent among cases versus controls, for most previously reported BrS-susceptibility genes” (Le Scouarnec et al., 2015).
Tester et al. (2012) examined 173 cases of sudden unexplained death that were referred for cardiac channel molecular autopsy. The mutational analysis included the long QT syndrome genes (KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2) and a catecholaminergic polymorphic ventricular tachycardia (CPVT) type 1–associated gene (RYR2). Overall, 45 putative pathogenic mutations absent in 400 to 700 controls were identified in 45 autopsy-negative SUD cases (26.0%) (Tester et al., 2012).
Seidelmann et al. (2017) evaluated the use of whole exome sequencing for clinical diagnosis, risk stratification, and management of inherited CVD. They found that genetic diagnosis was reached with a success rate of 26.5% (53/200 patients). This compares to 18% (36/200) that would have been diagnosed using commercially available genetic panels; although, this finding was not statistically significant. The authors concluded, “Whole exome sequencing was particularly useful for clinical diagnosis in patients with aborted sudden cardiac death, in whom the primary insult for the presence of both depressed cardiac function and prolonged QT had remained unknown. The analysis of the remaining cases using genome annotation and disease segregation led to the discovery of novel candidate genes in another 14% of the cases” (Seidelmann et al., 2017).
Munroe et al. (2018) examined tissue from 242 stillbirths (≥22 weeks), including those where no definite cause of death could be confirmed after a full autopsy. Seventy cases were examined, which were then sequenced for a custom panel of 35 genes. One putative pathogenic variant was found, and several novel variants of uncertain significance resulting in cardiac channelopathies was identified in some cases of otherwise unexplained stillbirth. The authors concluded “these variants may have a role in fetal demise” (Munroe et al., 2018).
Wang et al. (2017) examined the “genetic spectrum” of LVNC. The authors sequenced 73 cardiomyopathy-related genes in 102 patients, and 43 pathogenic variants were identified in 16 genes in 39 patients. Sarcomeric variants accounted for 63% of these variants whereas variants associated with channelopathies accounted for 12%. MYH7 and TAZ pathogenic variants were the most common, and patients with pathogenic variants showed more severe symptoms such as earlier age of onset (Wang et al., 2017).
van Lint et al. (2019) evaluated the detection rates for variants of unknown (class 3), likely (class 4), and certain (class 5) pathogenicity in cardiogenetic gene panels. The study included 936 patients that were evaluated with the arrhythmia panels (four versions), and 1970 patients were evaluated with the cardiomyopathy panels (six versions). The arrhythmia panels detected class 3 variants in 34.8% of patients, class 4 variants in 4.2% of patients, and class 5 variants in 4.6% of patients. The cardiomyopathy panels detected class 3 variants in 40.8% of patients, class 4 variants in 7.9% of patients, and class 5 variants in 12% of patients. Overall, the arrhythmia panels detected variants of interest in 44% of patients, and the cardiomyopathy panels detected variants of interest in 61% of patients. The authors concluded that “larger gene panels can increase the detection rate of likely pathogenic and pathogenic variants, but mainly increase the frequency of variants of unknown pathogenicity” (van Lint et al., 2019).
American Heart Association, American College of Cardiology, and the Heart Rhythm Society
In 2017 (Al-Khatib Sana et al., 2018) the American Heart Association (AHA), the American College of Cardiology (ACC), and the Heart Rhythm Society (HRS) published the Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death that recommends the following:
Genetic Considerations in Arrhythmia Syndromes
- In patients and family members in whom genetic testing for risk stratification for SCA or SCD is recommended, genetic counseling is beneficial. (I)
- The diagnosis of most inherited arrhythmia syndromes is based on clinical features and family history. The availability of genetic testing for inherited arrhythmia syndromes can: 1) provide opportunity to confirm a suspected clinical diagnosis and sometimes provide prognostic information for the proband and 2) offer cascade screening of potentially affected family members when a disease-causing mutation is identified in the proband. The yield of genetic testing varies by disease.
- Genotyping is frequently most useful when a pathogenic mutation is identified in the proband, such that screening can be applied to relatives who are in a preclinical phase, allowing institution of lifestyle changes, therapy, or ongoing monitoring for those who are gene mutation-positive.
- In young patients (<40 years of age) without structural heart disease who have unexplained cardiac arrest, unexplained near drowning, or recurrent exertional syncope, genetic testing may be important to identify an inherited arrhythmia syndrome as a likely cause.
Arrhythmogenic Right Ventricular Cardiomyopathy
- “Selected first-degree relatives refers to relatives who are willing to undergo further testing and who could benefit from further screening and testing (and not the terminally ill patients or those who do not want to be screened and tested).”
- “Arrhythmogenic right ventricular cardiomyopathy is detected clinically in approximately 35% to 40% of first-degree relatives, most commonly in siblings or symptomatic first-degree relatives”
- “The proband with arrhythmogenic right ventricular cardiomyopathy is usually diagnosed by the presence of clinical symptoms along with the presence of arrhythmogenic right ventricular cardiomyopathy Task Force criteria”
- “A pathogenic genetic mutation was added to the major Task Force criteria in 2010. The yield of genetic testing in probands with suspected arrhythmogenic right ventricular cardiomyopathy is generally 30% to 54% and is up to 58% among patients with a strong family history of SCD in multiple members. A negative genetic test for arrhythmogenic right ventricular cardiomyopathy does not exclude the disease, and a positive genetic test currently does not guide therapy.”
Hypertrophic Cardiomyopathy
- “In first-degree relatives of patients with HCM due to a known causative mutation, genetic counseling and mutation-specific genetic testing are recommended.”
- “In patients with clinically suspected or diagnosed HCM, genetic counseling and genetic testing are reasonable.”
- “When genetic testing reveals a mutation in the index patient, ascertainment of genetic status in first- and second-degree relatives can be predictive of risk for developing HCM. Relatives with overt HCM will have the same pathogenic HCM mutation as the index patient.”
Cardiac Channelopathies
- In first-degree relatives of patients who have a causative mutation for long QT syndrome, catecholaminergic polymorphic ventricular tachycardia, short QT syndrome, or Brugada syndrome, genetic counseling and mutation-specific genetic testing are recommended (I)
- Clinical screening of first-degree relatives of patients with inherited arrhythmia syndromes is crucial to identifying affected family members. Due to the increased risk of adverse cardiac events in genotype-positive patients with long QT syndrome, catecholaminergic polymorphic ventricular tachycardia, and Brugada syndrome, targeted screening for the identified family-specific mutation can identify individuals who are at risk for these adverse outcomes
Congenital Long QT Syndrome
- In patients with clinically diagnosed long QT syndrome, genetic counseling and genetic testing are recommended (I)
- Genetic testing for disease-causing mutations in long QT syndrome offers important diagnostic, prognostic, and therapeutic information in addition to the clinical evaluation, and a positive test can facilitate establishing risk for family members. The yield of genetic testing in long QT syndrome phenotype-positive patients is 50% to 86%, with the higher range present in patients with marked QT prolongation or positive family history of SCD. A negative genetic test does not exclude the diagnosis of long QT syndrome, which relies on the clinical evaluation.
Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
- In patients with catecholaminergic polymorphic ventricular tachycardia and with clinical VT or exertional syncope, genetic counseling and genetic testing are reasonable (IIa)
- Genetic testing may be useful to confirm the diagnosis of catecholaminergic polymorphic ventricular tachycardia, which is suggested by the development of bidirectional VT with exertion or stress. Recognition of catecholaminergic polymorphic ventricular tachycardia as the cause for exertional symptoms should prompt aggressive therapy to prevent the significant risk of SCD. Therapy for catecholaminergic polymorphic ventricular tachycardia is not guided by genotype status, but screening of first-degree relatives may be facilitated with genetic testing.
Brugada Syndrome
- In patients with suspected or established Brugada syndrome, genetic counseling and genetic testing may be useful to facilitate cascade screening of relatives (IIb)
- The yield of genetic testing in phenotype-positive patients is approximately 20% to 30% in Brugada syndrome. SCN5A variants account for most of this subset of genotype-positive Brugada syndrome. However, 2% to 10% of otherwise healthy individuals host a rare variant of SCN5A. A negative genetic test does not exclude the diagnosis of Brugada syndrome, which is usually based on electrocardiographic and clinical characteristics. Risk stratification is based on symptoms and clinical findings; genotype status is not correlated with the risk of adverse events. Identification of a pathogenetic mutation may help facilitate recognition of carrier status in family members, allowing for lifestyle modification and potential treatment.
Short QT syndrome
- In patients with short QT syndrome, genetic testing may be considered to facilitate screening of first-degree relatives (IIb)
- Pathogenic mutations in potassium channels have been identified in approximately 10% to 20% of patients with short QT syndrome, including in KCNH2 (SQT1), KCNQ1 (SQT2), and KCNJ2 (SQT3).
Early Repolarization “J-wave” Syndrome
- In patients with early repolarization pattern on ECG, genetic testing is not recommended (III-no benefit)
Postmortem Evaluation of SCD
- In first-degree relatives of SCD victims who were 40 years of age or younger, cardiac evaluation is recommended, with genetic counseling and genetic testing performed as indicated by clinical findings (I)
- For family risk profiling, it is important to use the disease-specific genetic test panel that corresponds to the autopsy findings. Risk profiling of family members of an SCD victim suspected of having an inherited cardiomyopathy at autopsy is important. Although phenotyping of surviving family members is crucial, genotyping of the SCD proband provides a mechanism for efficient follow-up evaluation of those relatives with the disease-causing mutation found in the proband (Al-Khatib Sana et al., 2018).
American Heart Association/American College of Cardiology/Heart Failure Society of America (AHA/ACC/HFSA) Guideline for the Management of Heart Failure
These 2022 guidelines provide guidelines on genetic evaluation and testing for the management of heart failure. The relevant recommendations are captured below:
- “In first-degree relatives of selected patients with genetic or inherited cardiomyopathies, genetic screening and counseling are recommended to detect cardiac disease and prompt consideration of treatments to decrease HF progression and sudden death.”
- “In select patients with nonischemic cardiomyopathy, referral for genetic counseling and testing is reasonable to identify conditions that could guide treatment for patients and family members.”
These joint recommendations also urge that “In patients in whom a genetic or inherited cardiomyopathy is suspected, a family history should be performed, including at least 3 generations and ideally diagrammed as a family tree pedigree” (Heidenreich et al., 2022).
American College of Cardiology Foundation (ACCF)/American College of Cardiology (ACC) and American Heart Association (AHA)
In 2020, the AHA released a joint guideline with the ACC for the diagnosis and treatment of patients with hypertrophic cardiomyopathy (Ommen et al., 2020). This guideline replaced the 2011 guideline released jointly by the ACCF and the AHA. The updated 2020 guideline has also been endorsed by the Heart Rhythm Society (HRS). With relation to genetic testing, they released the following with a level B-NR quality of evidence, signifying “moderate-quality evidence from 1 or more well-designed, well-executed nonrandomized studies, observational studies, or registry studies; meta-analyses of such studies”:
Class 1 (Benefit >>> Risk)
- “In patients with HCM, evaluation of familial inheritance, including a 3-generation family history, is recommended as part of the initial assessment
- In patients with HCM, genetic testing is beneficial to elucidate the genetic basis to facilitate the identification of family members at risk for developing HCM (cascade testing)
- In patients with an atypical clinical presentation of HCM or when another genetic condition is suspected to be the cause, a work-up including genetic testing for HCM and other genetic causes of unexplained cardiac hypertrophy (“HCM phenocopies”) is recommended
- In patients with HCM who choose to undergo genetic testing, pre- and post-test genetic counseling by an expert in the genetics of cardiovascular disease is recommended so that risks, benefits, results, and their clinical significance can be reviewed and discussed with the patient in a shared decision-making process
- When performing genetic testing in an HCM proband, the initial tier of genes tested should include genes with strong evidence to be disease-causing in HCM*
- In first-degree relatives of patients with HCM, both clinical screening (ECG and 2D echocardiogram) and cascade genetic testing (when a pathogenic/likely pathogenic variant has been identified in the proband) should be offered
- In families where a sudden unexplained death has occurred with a postmortem diagnosis of HCM, postmortem genetic testing is beneficial to facilitate cascade genetic testing and clinical screening in first-degree relatives
- In patients with HCM who have undergone genetic testing, serial reevaluation of the variant(s) identified is recommended to assess for variant reclassification, which may impact diagnosis and cascade genetic testing in family members
- In affected families with HCM, preconception and pre-natal reproductive and genetic counseling should be offered”
- NOTE: “Strong evidence HCM genes include, at the time of this publication, MYH7, MYBPC3, TNNI3, TNNT2, TPM1, MYL2, MYL3, and ACTC1”
Class 2b (Benefit ≥ Risk)
- “In patients with HCM, the usefulness of genetic testing in the assessment of risk of SCD [sudden cardiac death] is uncertain
- In patients with HCM who harbor a variant of uncertain significance, the usefulness of clinical genetic testing of phenotype-negative relatives for the purpose of variant reclassification is uncertain”
No benefit
- “For patients with HCM who have undergone genetic testing and were found to have no pathogenic variants (i.e., harbor only benign/likely benign variants), cascade genetic testing of the family is not useful
- Ongoing clinical screening is not indicated in genotype-negative relatives in families with genotype-positive HCM, unless the disease-causing variant is downgraded to variant of uncertain significance, likely benign, or benign variant during follow-up” (Ommen et al., 2020).
The ACCF and AHA also published a joint guideline, regarding the “Management of Heart Failure”, which states that “Increasingly, it is recognized that many (20% to 35%) patients with an idiopathic DCM have a familial cardiomyopathy (defined as 2 closely related family members who meet the criteria for idiopathic DCM).” “Advances in technology permitting high-throughput sequencing and genotyping at reduced costs have brought genetic screening to the clinical arena” and refers to the 2009 and 2011 published guidelines. The guidelines further note that genetic testing may be considered in conjunction with counseling in familial DCM (Yancy et al., 2013).
American Heart Association (AHA)
The AHA published a guideline regarding cardiomyopathy in children. In it, they state that “genetic testing should first be performed in the individual known to have a specific cardiomyopathy phenotype and should be informed by the child’s overall presentation, with a detailed examination looking for dysmorphic features, muscle weakness, scoliosis, or specific laboratory findings.” They also state that indications for genetic testing include “determining the cause of HCM, predicting the clinical course and severity, screening first-degree relatives, and determining recurrence risk.”
This guideline addresses HCM, DCM, RCM, LVNC, Arrhythmogenic Ventricular Cardiomyopathy (both right and left ventricles), as well as cardiomyopathy caused by channelopathies such as Long-QT syndrome (Lipshultz et al., 2019). However, though it addresses the many manifestations of cardiomyopathy, few suggestions for clinical practice are made due to “the lack of evidence and consensus in the classification and diagnosis of children with cardiomyopathy,” such that the document takes the form of a scientific statement instead of clinical guidelines.
In a 2023 scientific statement regarding cardiomyopathy in children, the AHA goes on to state that when “cardiomyopathy is identified in a child without a known preexisting risk, panel genetic testing or whole exome sequencing is recommended. A pathogenic or likely pathogenic variant in the proband should prompt cascade genetic testing of first-degree relatives who are at risk for cardiomyopathy” (Bogle et al., 2023).
The AHA also published a guideline discussing early repolarization. In it, they remark that the biological basis for early repolarization pattern “remains incompletely understood.” Therefore, the AHA proposes “large-scale, unbiased (e.g., genome-wide association studies), family-guided genetic discovery approaches, as well as efforts aimed at understanding both the mechanisms underlying ER and the associated arrhythmogenesis” (Patton et al., 2016).
In a 2020 statement from the AHA titled “Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement from the American Heart Association”, the AHA pointed towards a 2018 conjoint guideline by the HFSA with the ACMG for guidance for genetic testing of cardiomyopathies. The pertinent recommendations were extracted and are presented below:
“A family history of at least 3 generations should be obtained for all patients with a primary cardiomyopathy. Second, clinical screening for cardiomyopathy is recommended for at-risk first-degree relatives. Third, patients with genetic, familial, or other unexplained forms of cardiomyopathy should be referred to expert centers. Genetic counseling is recommended for all patients with cardiomyopathy and their family members.
In a family, testing should be directed to the most clearly affected family member. If that individual is found to have a gene variant that is judged to be pathogenic or likely pathogenic, then cascade genetic testing for that variant should be offered to at-risk family members. For infants with cardiomyopathy, in addition to any routine newborn screening tests that might have been performed, the specialized evaluation is likely to include genetic testing and should also include an evaluation for syndromic or metabolic conditions for which a specific intervention or therapy might be warranted” (Musunuru et al., 2020).
Finally, the guideline notes that the management of certain conditions may benefit or otherwise be influenced by the results of genetic testing. Long-QT syndrome, HCM, DCM, and RCM are listed as having variants that may be specifically targeted with certain treatments (Musunuru et al., 2020).
Heart Failure Society of American and American College of Medical Genetics and Genomics (ACMG)
In 2018, the Heart Failure Society of America and the American College of Medical Genetics and Genomics published a clinical practice resource for the genetic evaluation of cardiomyopathy. There,
“Recommendation 1. Genetic testing is recommended for patients with cardiomyopathy
- Genetic testing is recommended for the most clearly affected family member.
- Cascade genetic testing of at-risk family members is recommended for pathogenic and likely pathogenic variants.
- In addition to routine newborn screening tests, specialized evaluation of infants with cardiomyopathy is recommended, and genetic testing should be considered.”
On the point of whom to test, the societies have the following to say:
“To yield the most conclusive, informative results, diagnostic genetic testing is optimally initiated on a confirmed affected individual. Furthermore, as there are sometimes multiple genetic variants contributing to disease in a single family, the testing should ideally be initiated on the person who is most likely to harbor the disease-causing variant or variants. This is frequently the individual in the family with the most severe disease and/or the earliest disease onset. This is a well established principle in clinical genetics, as selecting the individual with the most evident disease increases the likelihood of finding a genetic cause. If the ideal person for initiation of genetic testing in a family is unavailable or unwilling to proceed, then comprehensive genetic testing should be considered for another unequivocally affected family member.”
As for when to test, ACMG “recommend[s] genetic testing at the time a new cardiomyopathy diagnosis is made, but it can be conducted at any time following diagnosis. Education and counseling regarding genetic testing options are a key component of the process. For those who have had genetic testing in the past, retesting may be appropriate if the previous testing produced negative or inconclusive results and the test’s detection rate has improved. This latter point is particularly relevant for DCM as the gene panels have rapidly expanded (e.g., TTN and others) and are anticipated to continue” (Hershberger et al., 2018). A chart of selected genes implicated in various cardiomyopathies is included below.
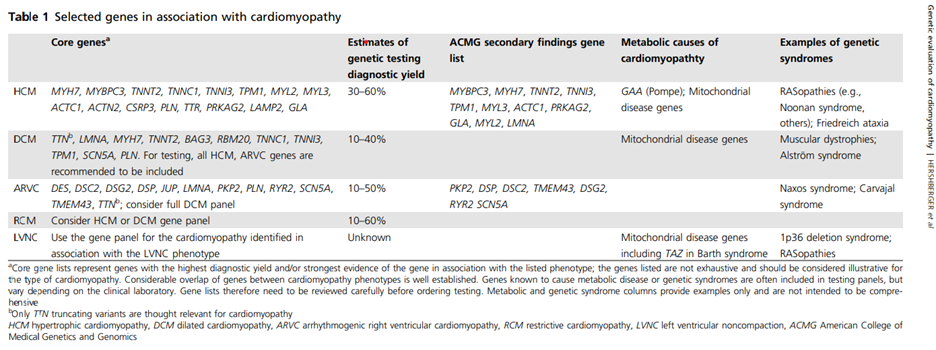
(Table 1 above appropriated from (Hershberger et al., 2018))
The specific cardiomyopathy comments are as follows:
HCM
- “Beyond sarcomeric genes, core genes to screen in patients with HCM include GLA, PRKAG2, and LAMP2.”
- “Consultation with a geneticist is indicated.”
DCM
- “Core genes to be tested in individuals with DCM include genes encoding sarcomeric and cytoskeletal proteins.”
- “Genetic testing is important in mothers of individuals with Duchenne or Becker to determine carrier status because carrier females may develop DCM in the third to fifth decade of life.”
LVNC
- “Genetic testing is not recommended when the LVNC phenotype is identified serendipitously in asymptomatic individuals with otherwise normal cardiovascular structure and function.”
As such, these genes may form an important backdrop upon which the following is performed:
“Recommendation 2. Focused cardiovascular phenotyping is recommended when pathogenic or likely pathogenic variants in cardiomyopathy genes, designated for reporting of secondary findings by the ACMG, are identified in an individual.
- If a cardiovascular phenotype is identified as would be predicted by currently available knowledge of the gene/ variant pair, all usual approaches described in this document for a genetic evaluation, including family-based approaches, are recommended.
- If no cardiovascular disease phenotype is identified in the individual, recommendations for surveillance screening at intervals should be considered.
- If no cardiovascular phenotype is identified in the individual, cascade evaluation of at-risk relatives may be considered, tempered by the strength of evidence supporting the pathogenicity of the variant, the usual age of onset of the gene/variant pair, and pedigree information (e.g., the ages of at-risk family members, other previously known cardiovascular clinical data in the pedigree, and related information)” (Hershberger et al., 2018).
American College of Medical Genetics and Genomics (ACMG)
In 2023 version 3.2, ACMG referenced several myopathies and channelopathies in their “minimum” list of genes for which mutations should be reported when whole genome sequencing is performed for other primary purposes (incidental findings). Those genes are listed below:
- FBN1, TGFBR1, TGFBR2, SMAD3, ACTA2, MYH11 for aortopathies
- PKP2, DSP, DSC2, TMEM43, and DSG2 for ARVC
- RYR2, CASQ2, and TRDN for Catecholaminergic polymorphic ventricular tachycardia (CPVT)
- TNNT2, LMNA, FLNC, TTN, BAG3, DES, RBM20, and TNNC1 for DCM
- COL3A1 for Ehlers-Danlos syndrome, vascular type
- LDLR, APOB, and PCSK9 for Familial hypercholesterolemia
- MYH7, MYBP3, TNNI3, TPM1, MYL3, ACTC1, PRKAG2, and MYL2 for HCM
- KCNQ1 and KCNH2 for Long QT syndrome types 1 and 2
- SCN5A for Long QT syndrome 3; Brugada syndrome
- CALM1, CALM2, CALM3 for Long QT syndrome types 14-16 (Miller et al., 2023).
In an erratum published in 2021, the authors appended a series of new gene-phenotype pairings, each associated with the cardiovascular phenotype “Risk of sudden death with preventive interventions available”: CASQ2/catecholaminergic polymorphic ventricular tachycardia (CPVT), TRDN/catecholaminergic polymorphic ventricular tachycardia (CPVT) & long QT syndrome, FLNC/cardiomyopathy, and TTN/cardiomyopathy (Miller et al., 2021).
The Heart Rhythm Society (HRS)/The European Heart Rhythm Association (EHRA) Expert Consensus Statement
Hypertrophic Cardiomyopathy (HCM)
Class I recommendations (is recommended):
- “Comprehensive or targeted (MYBPC3, MYH7, TNNI3, TNNT2, TPM1) HCM genetic testing is recommended for any patient in whom a cardiologist has established a clinical diagnosis of HCM based on examination of the patient's clinical history, family history, and electrocardiographic/echocardiographic phenotype.”
- “Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the HCM-causative mutation in an index case.”
Dilated Cardiomyopathy (DCM)
Class I recommendations:
- “Comprehensive or targeted (LMNA and SCN5A) DCM genetic testing is recommended for patients with DCM and significant cardiac conduction disease (i.e., first-, second-, or third- degree heart block) and/or with a family history of premature unexpected sudden death.”
- “Mutation-specific testing is recommended for family members and appropriate relatives following the identification of a DCM-causative mutation in the index case.”
Class IIa (can be useful) recommendations:
- Genetic testing can be useful for patients with familial DCM to confirm the diagnosis, to recognize those who are highest risk of arrhythmia and syndromic features, to facilitate cascade screening within the family, and to help with family planning.
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
Class I recommendations:
- “Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the ACM/ARVC- causative mutation in an index case.”
Class II recommendations:
- “Comprehensive or targeted (DSC2, DSG2, DSP, JUP, PKP2, and TMEM43) ACM/ARVC genetic testing can be useful for patients satisfying task force diagnostic criteria for ACM/ARVC.” (II-A, can be useful)
- “Genetic testing may be considered for patients with possible ACM/ARVC (1 major or 2 minor criteria) according to the 2010 task force criteria (European Heart Journal).” (II-B, may be considered)
Left Ventricular Noncompaction (LVNC)
Class I recommendations:
- “Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of a LVNC-causative mutation in the index case.”
Class IIa recommendations:
- LVNC genetic testing can be useful for patients in whom a cardiologist has established a clinical diagnosis of LVNC based on examination of the patient's clinical history, family history, and electrocardiographic/echocardiographic phenotype. (II-A)
Restrictive Cardiomyopathy (RCM)
Class I recommendations:
- “Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of a RCM-causative mutation in the index case.”
Class IIb (may be considered) recommendations:
- “RCM genetic testing may be considered for patients in whom a cardiologist has established a clinical index of suspicion for RCM based on examination of the patient's clinical history, family history, and electrocardiographic/echocardiographic phenotype.”
Long-QT Syndrome
Class I recommendations:
- “Comprehensive or LQT1-3 (KCNQ1, KCNH2, and SCN5A) targeted LQTS genetic testing is recommended for any patient in whom a cardiologist has established a strong clinical index of suspicion for LQTS based on examination of the patient’s clinical history, family history, and expressed electrocardiographic (resting 12-lead ECGs and/or provocative stress testing with exercise or catecholamine infusion) phenotype.
- Comprehensive or LQT1-3 (KCNQ1, KCNH2, and SCN5A) targeted LQTS genetic testing is recommended for any asymptomatic patient with QT prolongation in the absence of other clinical conditions that might prolong the QT interval (such as electrolyte abnormalities, hypertrophy, bundle branch block, etc., i.e., otherwise idiopathic) on serial 12-lead ECGs defined as QTc >480 ms (prepuberty) or >500 ms (adults).
- Mutation-specific genetic testing is recommended for family members and other appropriate relatives subsequently following the identification of the LQTS-causative mutation in an index case.”
Class IIb recommendations:
- “Comprehensive or LQT1-3 (KCNQ1, KCNH2, and SCN5A) targeted LQTS genetic testing may be considered for any asymptomatic patient with otherwise idiopathic QTc values >460 ms (prepuberty) or >480 ms (adults) on serial 12-lead ECGs.”
Catecholaminergic polymorphic ventricular tachycardia (CPVT)
Class I recommendations:
- Comprehensive or CPVT1 and CVPT2 (RYR2 and CASQ2) targeted CPVT genetic testing is recommended for any patient in whom a cardiologist has established a clinical index of suspicion for CPVT based on examination of the patient's clinical history, family history, and expressed electrocardiographic phenotype during provocative stress testing with cycle, treadmill, or catecholamine infusion.
- Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the CPVT-causative mutation in an index case.
Brugada Syndrome
Class I recommendations:
- Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the BrS-causative mutation in an index case.
Class IIa recommendations:
- Comprehensive or BrS1 (SCN5A) targeted BrS genetic testing can be useful for any patient in whom a cardiologist has established a clinical index of suspicion for BrS based on examination of the patient's clinical history, family history, and expressed electrocardiographic (resting 12-lead ECGs and/or provocative drug challenge testing) phenotype
Genetic testing is not indicated in the setting of an isolated type 2 or type 3 Brugada ECG pattern.
Short QT syndrome
Class I recommendations:
- Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the SQTS-causative mutation in an index case.
Class IIb recommendations:
- Comprehensive or SQT1-3 (KCNH2, KCNQ1, and KCNJ2) targeted SQTS genetic testing may be considered for any patient in whom a cardiologist has established a strong clinical index of suspicion for SQTS based on examination of the patient's clinical history, family history, and electrocardiographic phenotype.
Sinus Node Dysfunction (SND)
- “In idiopathic sinus node dysfunction (SND), mutations in the cardiac pacemaker channel gene HCN4 and in sodium channel genes can be identified in an unknown portion. However, because non-genetic causes appear more frequently in idiopathic SND, genetic testing for idiopathic SND should be considered on an individual basis”.
Progressive Cardiac Conduction Disease (CCD)
- “Genetic testing may be considered as part of the diagnostic evaluation for patients with either isolated CCD or CCD with concomitant congenital heart disease, especially when there is documentation of a positive family history of CCD.”
- “Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the CCD-causative mutation in an index case.”
- “Taken together, tiered genetic testing for patients with CCD and congenital heart disease or cardiomyopathies is useful because other cardiac and non-cardiac disease features may be present or may develop; individual genes should be considered after discussion of clinical features with an experienced cardiogenetic center”.
Post-Mortem Genetic Testing in Sudden Unexpected Death Cases
- “In the setting of autopsy-negative SUDS, comprehensive or targeted (RYR2, KCNQ1, KCNH2, and SCN5A) ion channel genetic testing may be considered in an attempt to establish probable cause and manner of death and to facilitate the identification of potentially at-risk relatives and is recommended if circumstantial evidence points toward a clinical diagnosis of LQTS or CPVT specifically (such as emotional stress, acoustic trigger, drowning as the trigger of death).”
- “Mutation-specific genetic testing is recommended for family members and other appropriate relatives following the identification of a SUDS-causative mutation in the decedent” (Ackerman et al., 2011; M. J. Ackerman et al., 2011).
Heart Rhythm Society (HRS), European Heart Rhythm Association (EHRA), and Asia Pacific Heart Rhythm Society (APHRS)
In an international consensus statement released by the HRS, EHRA, and APHRS, the three medical societies provide expert recommendations for diagnosing “inherited primary arrhythmia syndromes.” The pertinent guidelines are listed below:
- “Expert Consensus Recommendations on LQTS Diagnosis
- LQTS is diagnosed:
- In the presence of an LQTS risk score ≥3.5 in the absence of a secondary cause for QT prolongation and/or
- In the presence of an unequivocally pathogenic mutation in one of the LQTS genes or
- In the presence of a QT interval corrected for heart rate using Bazett’s formula (QTc) ≥500 ms in repeated 12-lead electrocardiogram (ECG) and in the absence of a secondary cause for QT prolongation.
- LQTS can be diagnosed in the presence of a QTc between 480-499 ms in repeated 12-lead ECGs in a patient with unexplained syncope in the absence of a secondary cause for QT prolongation and in the absence of a pathogenic mutation.”
- LQTS is diagnosed:
- “Expert Consensus Recommendations on Brugada Syndrome Diagnosis
- BrS is diagnosed in patients with ST-segment elevation with type 1 morphology ≥2 mm in ≥1 lead among the right precordial leads V1, V2, positioned in the 2nd, 3rd or 4th intercostal space occurring either spontaneously or after provocative drug test with intravenous administration of Class I antiarrhythmic drugs.
- BRS is diagnosed in patients with type 2 or type 3 ST-segment elevation in ≥1 lead among the right precordial leads V1, V2 positioned in the 2nd, 3rd or 4th intercostal space when a provocative test with intravenous administration of Class I antiarrhythmic drugs induces a type I ECG morphology.”
- “Expert Consensus Recommendations on [Catecholaminergic Polymorphic Ventricular Tachycardia] Diagnosis
- CPVT is diagnosed in the presence of a structurally normal heart, normal ECG, and unexplained exercise or catecholamine-induced bidirectional VT or polymorphic ventricular premature beats or VT in an individual < 40 years of age.
- CPVT is diagnosed in patients (index case or family member) who have a pathogenic mutation.
- CPVT is diagnosed in family members of a CPVT index case with a normal heart who manifest exercise-induced premature ventricular contractions (PVCs) or bidirectional/ polymorphic VT.
- CPVT can be diagnosed in the presence of a structurally normal heart and coronary arteries, normal ECG, and unexplained exercise or catecholamine-induced bidirectional VT or polymorphic ventricular premature beats or VT in an individual >40 years of age.”
- “Expert Consensus Recommendations on Short QT Syndrome Diagnosis
- SQTS is diagnosed in the presence of a QTc ≤330 ms.
- SQTS can be diagnosed in the presence of a QTc <360 ms and one or more of the following: a pathogenic mutation, family history of SQTS, family history of sudden death at age ≤40, survival of a VT/VF episode in the absence of heart disease.”
- “Expert Consensus Recommendations on Early Repolarization Diagnosis
- ER syndrome is diagnosed in the presence of J-point elevation ≥1mm in ≥2 contiguous inferior and/or lateral leads of a standard 12-lead ECG in a patient resuscitated from otherwise unexplained VF/polymorphic VT.
- ER syndrome can be diagnosed in an SCD victim with a negative autopsy and medical chart review with a previous ECG demonstrating J-point elevation ≥1mm in ≥2 contiguous inferior and/or lateral leads of a standard 12-lead ECG
- ER syndrome can be diagnosed in the presence of J-point elevation ≥1mm in ≥2 contiguous inferior and/or lateral leads of a standard 12-lead ECG.”
- “Expert Consensus Recommendations on Progressive Cardiac Conduction Disease Diagnosis
- Progressive cardiac conduction disease (PCCD) is diagnosed in the presence of unexplained progressive conduction abnormalities in young (<50 years) individuals with structurally normal hearts in the absence of skeletal myopathies, especially if there is a family history of PCCD.”
- “Unexplained Cardiac Arrest:
- Expert Consensus Recommendations on Idiopathic Ventricular Fibrillation (IVF) Diagnosis
- IVF is defined as a resuscitated cardiac arrest victim, preferably with documentation of VF, in whom known cardiac, respiratory, metabolic and toxicological etiologies have been excluded through clinical evaluation.
- Expert Consensus Recommendations on Idiopathic Ventricular Fibrillation Evaluation
- Class IIa - Genetic testing in IVF can be useful when there is a suspicion of a specific genetic disease following clinical evaluation of the IVF patient and/or family members.
- Class III - Genetic screening of a large panel of genes in IVF patients in whom there is no suspicion of an inherited arrhythmogenic disease after clinical evaluation should not be performed.
- Expert Consensus Recommendations on Idiopathic Ventricular Fibrillation (IVF) Diagnosis
- “Unexplained Sudden Cardiac Death: Expert Consensus Recommendations on Sudden Unexplained Death Syndrome Evaluation
- Class I – Collection of blood and/or suitable tissue for molecular autopsy/postmortem genetic testing is recommended in all SUDS victims.
- Class IIa – An arrhythmia syndrome focused molecular autopsy/postmortem genetic testing can be useful for all SUDS victims” (Priori et al., 2013)
Heart Rhythm Society (HRS)
Arrhythmogenic Cardiomyopathies
The HRS published an “Expert Consensus Statement” on “Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy.” In it, they include recommendations on genetic testing for this set of cardiac disorders.
“For individuals and decedents with either a clinical or necropsy diagnosis of ACM, genetic testing of the established ACM-susceptibility genes is recommended.”
“For genetic testing of the established ACM-susceptibility genes, comprehensive analysis of all established genes with full coverage is recommended.”
“When a likely pathogenic or pathogenic genetic variant has been identified in the proband, cascade genetic testing can be offered to first-degree at-risk relatives… Cascade genetic testing is therefore only offered to family members where a likely pathogenic or pathogenic variant in a known disease-associated gene is identified in the proband…” (Towbin et al., 2019).
Canadian Cardiovascular Society/Canadian Heart Rhythm Society
Long QT Syndrome
Genetic testing is recommended in the cardiac arrest survivor with LQTS for the primary purpose of screening first-degree relatives, in the patient with syncope and QTc prolongation that is attributed to LQTS, and in the asymptomatic patient with consistent QTc prolongation that is clinically suspected to represent LQTS.
Brugada Syndrome
Genetic testing in the cardiac arrest survivor, patient with syncope, or asymptomatic patient with a persistent or provocable type 1 Brugada ECG pattern is recommended for the primary purpose of screening of family members. However, patients with types 2 or 3 Brugada ECG patterns are not recommended for genetic testing.
European Society of Cardiology (ECS)
Genetic counselling is recommended for all HCM patients when the HCM is not explained solely by a non-genetic cause.
Genetic testing is recommended for patients fulfilling the diagnostic criteria for HCM, both as a confirmatory test and to enable genetic testing for relatives. Both cascade genetic screening and a clinical evaluation are recommended for first-degree relatives that carry the same mutation as the HCM patient (“proband”). Even if a mutation is absent, relatives should consider reassessment should symptoms appear, or other clinical data emerges. A genetic analysis, such as pedigree analysis and high-throughput sequencing, should include the most implicated sarcomere protein genes. If a rarer condition is suspected, the analysis should include the gene responsible for that condition.
Pre-natal genetic testing is not recommended due to phenotypic variability (Elliott et al., 2014).
In 2023, the ESC released clinical practice guidelines delineating the management of cardiomyopathies. The pertinent recommendations from this update are listed below:
References
- Ackerman, Hamilton, R., Hershberger, R. E., Le Marec, H., McKenna, W. J., Schulze-Bahr, E., Semsarian, C., Towbin, J. A., Watkins, H., Wilde, A., Priori, S. G., Wolpert, C., Zipes, D. P., Willems, S., Berul, C., Brugada, R., Calkins, H., Judge, D. P., Camm, A. J., . . . Gollob, M. (2011). HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). EP Europace, 13(8), 1077-1109. https://doi.org/10.1093/europace/eur245
- Ackerman, M. J., Priori, S. G., Willems, S., Berul, C., Brugada, R., Calkins, H., Camm, A. J., Ellinor, P. T., Gollob, M., Hamilton, R., Hershberger, R. E., Judge, D. P., Le Marec, H., McKenna, W. J., Schulze-Bahr, E., Semsarian, C., Towbin, J. A., Watkins, H., Wilde, A., . . . Zipes, D. P. (2011). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm, 8(8), 1308-1339. https://doi.org/10.1016/j.hrthm.2011.05.020 AHA. (2024, May 28). Dilated Cardiomyopathy (DCM). https://www.heart.org/en/health-topics/cardiomyopathy/what-is-cardiomyopathy-in-adults/dilated-cardiomyopathy-dcm
- Al-Khatib Sana, M., Stevenson William, G., Ackerman Michael, J., Bryant William, J., Callans David, J., Curtis Anne, B., Deal Barbara, J., Dickfeld, T., Field Michael, E., Fonarow Gregg, C., Gillis Anne, M., Granger Christopher, B., Hammill Stephen, C., Hlatky Mark, A., Joglar José, A., Kay, G. N., Matlock Daniel, D., Myerburg Robert, J., & Page Richard, L. (2018). 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation, 138(13), e272-e391. https://doi.org/10.1161/CIR.0000000000000549
- Alcalai, R., Seidman, J. G., & Seidman, C. E. (2008). Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol, 19(1), 104-110. https://doi.org/10.1111/j.1540-8167.2007.00965.x
- Ammash, N. M. (2024, January 10). Restrictive cardiomyopathies. https://www.uptodate.com/contents/restrictive-cardiomyopathies
- Arbelo, E., Protonotarios, A., Gimeno, J. R., Arbustini, E., Barriales-Villa, R., Basso, C., Bezzina, C. R., Biagini, E., Blom, N. A., de Boer, R. A., De Winter, T., Elliott, P. M., Flather, M., Garcia-Pavia, P., Haugaa, K. H., Ingles, J., Jurcut, R. O., Klaassen, S., Limongelli, G., . . . Kaski, J. P. (2023). 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J, 44(37), 3503-3626. https://doi.org/10.1093/eurheartj/ehad194
- Attenhofer-Jost, C., & Connolly, H. (2022, May 5). Isolated left ventricular noncompaction in adults: Clinical manifestations and diagnosis. https://www.uptodate.com/contents/isolated-left-ventricular-noncompaction-in-adults-clinical-manifestations-and-diagnosis
- Bastiaenen, R., & Behr, E. R. (2011). Sudden death and ion channel disease: pathophysiology and implications for management. Heart, 97(17), 1365-1372. https://doi.org/10.1136/hrt.2011.223883
- Bezzina, C. R., Lahrouchi, N., & Priori, S. G. (2015). Genetics of Sudden Cardiac Death. https://doi.org/10.1161/CIRCRESAHA.116.304030
- Bhonsale, A., Groeneweg, J. A., James, C. A., Dooijes, D., Tichnell, C., Jongbloed, J. D., Murray, B., te Riele, A. S., van den Berg, M. P., Bikker, H., Atsma, D. E., de Groot, N. M., Houweling, A. C., van der Heijden, J. F., Russell, S. D., Doevendans, P. A., van Veen, T. A., Tandri, H., Wilde, A. A., . . . Hauer, R. N. (2015). Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J, 36(14), 847-855. https://doi.org/10.1093/eurheartj/ehu509
- BluePrint. (2024). Comprehensive Cardiology Panel. Retrieved February 19 from https://blueprintgenetics.com/tests/panels/cardiology/comprehensive-cardiology-panel/
- Bogle, C., Colan, S. D., Miyamoto, S. D., Choudhry, S., Baez-Hernandez, N., Brickler, M. M., Feingold, B., Lal, A. K., Lee, T. M., Canter, C. E., Lipshultz, S. E., on behalf of the American Heart Association Young Hearts Pediatric Heart, F., Transplantation Committee of the Council on Lifelong Congenital Heart, D., & Heart Health in the, Y. (2023). Treatment Strategies for Cardiomyopathy in Children: A Scientific Statement From the American Heart Association. Circulation, 148(2), 174-195. https://doi.org/10.1161/CIR.0000000000001151
- Campuzano, O., Sarquella-Brugada, G., Brugada, R., & Brugada, J. (2015). Genetics of channelopathies associated with sudden cardiac death. Glob Cardiol Sci Pract, 2015(3), 39. https://doi.org/10.5339/gcsp.2015.39
- Cirino, A. L., Ho, Carolyn. (2014). Hypertrophic Cardiomyopathy Overview [Text]. https://www.ncbi.nlm.nih.gov/books/NBK1768/
- Cirino, A. L., Lakdawala, N. K., McDonough, B., Conner, L., Adler, D., Weinfeld, M., O'Gara, P., Rehm, H. L., Machini, K., Lebo, M., Blout, C., Green, R. C., MacRae, C. A., Seidman, C. E., & Ho, C. Y. (2017). A Comparison of Whole Genome Sequencing to Multigene Panel Testing in Hypertrophic Cardiomyopathy Patients. Circ Cardiovasc Genet, 10(5). https://doi.org/10.1161/circgenetics.117.001768
- Cooper Jr, L. T. (2022, August 16). Definition and classification of the cardiomyopathies. https://www.uptodate.com/contents/definition-and-classification-of-the-cardiomyopathies
- Elliott, P. M., Anastasakis, A., Borger, M. A., Borggrefe, M., Cecchi, F., Charron, P., Hagege, A. A., Lafont, A., Limongelli, G., Mahrholdt, H., McKenna, W. J., Mogensen, J., Nihoyannopoulos, P., Nistri, S., Pieper, P. G., Pieske, B., Rapezzi, C., Rutten, F. H., Tillmanns, C., & Watkins, H. (2014). 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of
- Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J, 35(39), 2733-2779. https://doi.org/10.1093/eurheartj/ehu284
- Fernandez-Falgueras, A., Sarquella-Brugada, G., Brugada, J., Brugada, R., & Campuzano, O. (2017). Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology (Basel), 6(1). https://doi.org/10.3390/biology6010007
- Frustaci, A., De Luca, A., Guida, V., Biagini, T., Mazza, T., Gaudio, C., Letizia, C., Russo, M. A., Galea, N., & Chimenti, C. (2018). Novel alpha-Actin Gene Mutation p.(Ala21Val) Causing Familial Hypertrophic Cardiomyopathy, Myocardial Noncompaction, and Transmural Crypts. Clinical-Pathologic Correlation. J Am Heart Assoc, 7(4). https://doi.org/10.1161/jaha.117.008068
- Fulgent. (2024). Hypertrophic Cardiomyopathy NGS Panel. https://www.fulgentgenetics.com/hypertrophic-cardiomyopathy
- Garcia-Elias, A., & Benito, B. (2018). Ion Channel Disorders and Sudden Cardiac Death [Review]. International Journal of Molecular Sciences, 19(3), 692. https://doi.org/10.3390/ijms19030692
- Garcia, J., Tahiliani, J., Johnson, N. M., Aguilar, S., Beltran, D., Daly, A., Decker, E., Haverfield, E., Herrera, B., Murillo, L., Nykamp, K., & Topper, S. (2016). Clinical Genetic Testing for the Cardiomyopathies and Arrhythmias: A Systematic Framework for Establishing Clinical Validity and Addressing Genotypic and Phenotypic Heterogeneity. Front Cardiovasc Med, 3, 20. https://doi.org/10.3389/fcvm.2016.00020 GeneDX. (2024). GeneDx Testing Directory. Retrieved 1/14/21 from https://www.genedx.com/
- Gersh, B. J., Maron, B. J., Bonow, R. O., Dearani, J. A., Fifer, M. A., Link, M. S., Naidu, S. S., Nishimura, R. A., Ommen, S. R., Rakowski, H., Seidman, C. E., Towbin, J. A., Udelson, J. E., & Yancy, C. W. (2011). 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg, 142(6), e153-203. https://doi.org/10.1016/j.jtcvs.2011.10.020
- Giudicessi, J. R., & Ackerman, M. J. (2013). Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res, 161(1), 1-14. https://doi.org/10.1016/j.trsl.2012.08.005
- Gollob, M. H., Blier, L., Brugada, R., Champagne, J., Chauhan, V., Connors, S., Gardner, M., Green, M. S., Gow, R., Hamilton, R., Harris, L., Healey, J. S., Hodgkinson, K., Honeywell, C., Kantoch, M., Kirsh, J., Krahn, A., Mullen, M., Parkash, R., . . . Woo, A. (2011). Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society joint position paper. Can J Cardiol, 27(2), 232-245. https://doi.org/10.1016/j.cjca.2010.12.078
- Grondin, S., Davies, B., Cadrin-Tourigny, J., Steinberg, C., Cheung, C. C., Jorda, P., Healey, J. S., Green, M. S., Sanatani, S., Alqarawi, W., Angaran, P., Arbour, L., Antiperovitch, P., Khan, H., Leather, R., Guerra, P. G., Rivard, L., Simpson, C. S., Gardner, M., . . . Tadros, R. (2022). Importance of genetic testing in unexplained cardiac arrest. Eur Heart J, 43(32), 3071-3081. https://doi.org/10.1093/eurheartj/ehac145
- Heidenreich, P. A., Bozkurt, B., Aguilar, D., Allen, L. A., Byun, J. J., Colvin, M. M., Deswal, A., Drazner, M. H., Dunlay, S. M., Evers, L. R., Fang, J. C., Fedson, S. E., Fonarow, G. C., Hayek, S. S., Hernandez, A. F., Khazanie, P., Kittleson, M. M., Lee, C. S., Link, M. S., . . . Yancy, C. W. (2022). 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation, 145(18), e895-e1032. https://doi.org/10.1161/cir.0000000000001063
- Hershberger, R. E. (2023, April 17). Familial dilated cardiomyopathy: Prevalence, diagnosis and treatment. https://www.uptodate.com/contents/familial-dilated-cardiomyopathy-prevalence-diagnosis-and-treatment
- Hershberger, R. E., Givertz, M. M., Ho, C. Y., Judge, D. P., Kantor, P. F., McBride, K. L., Morales, A., Taylor, M. R. G., Vatta, M., & Ware, S. M. (2018). Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. https://doi.org/10.1038/s41436-018-0039-z
- Hershberger, R. E., Lindenfeld, J., Mestroni, L., Seidman, C. E., Taylor, M. R., & Towbin, J. A. (2009). Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail, 15(2), 83-97. https://doi.org/10.1016/j.cardfail.2009.01.006
- Hershberger, R. E., Morales, A., & Siegfried, J. D. (2010). Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med, 12(11), 655-667. https://doi.org/10.1097/GIM.0b013e3181f2481f
- Hershberger, R. E., & Siegfried, J. D. (2011). Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol, 57(16), 1641-1649. https://doi.org/10.1016/j.jacc.2011.01.015
- Hofman, N., Tan, H. L., Alders, M., Kolder, I., de Haij, S., Mannens, M. M., Lombardi, M. P., Dit Deprez, R. H., van Langen, I., & Wilde, A. A. (2013). Yield of molecular and clinical testing for arrhythmia syndromes: report of 15 years' experience. Circulation, 128(14), 1513-1521. https://doi.org/10.1161/circulationaha.112.000091
- Invitae. (2024). Invitae Cardiomyopathy Comprehensive Panel. https://www.invitae.com/en/physician/tests/02251/
- Japp, A. G., Gulati, A., Cook, S. A., Cowie, M. R., & Prasad, S. K. (2016). The Diagnosis and Evaluation of Dilated Cardiomyopathy. J Am Coll Cardiol, 67(25), 2996-3010. https://doi.org/10.1016/j.jacc.2016.03.590
- Kayvanpour, E., Sedaghat-Hamedani, F., Amr, A., Lai, A., Haas, J., Holzer, D. B., Frese, K. S., Keller, A., Jensen, K., Katus, H. A., & Meder, B. (2017). Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol, 106(2), 127-139. https://doi.org/10.1007/s00392-016-1033-6
- Kirk, J. A., & Kass, D. A. (2015). Cellular and Molecular Aspects of Dyssynchrony and Resynchronization. Card Electrophysiol Clin, 7(4), 585-597. https://doi.org/10.1016/j.ccep.2015.08.011
- Kostareva, A., Kiselev, A., Gudkova, A., Frishman, G., Ruepp, A., Frishman, D., Smolina, N., Tarnovskaya, S., Nilsson, D., Zlotina, A., Khodyuchenko, T., Vershinina, T., Pervunina, T., Klyushina, A., Kozlenok, A., Sjoberg, G., Golovljova, I., Sejersen, T., & Shlyakhto, E. (2016). Genetic Spectrum of Idiopathic Restrictive Cardiomyopathy Uncovered by Next-Generation Sequencing. PLoS One, 11(9), e0163362. https://doi.org/10.1371/journal.pone.0163362
- Le Scouarnec, S., Karakachoff, M., Gourraud, J. B., Lindenbaum, P., Bonnaud, S., Portero, V., Duboscq-Bidot, L., Daumy, X., Simonet, F., Teusan, R., Baron, E., Violleau, J., Persyn, E., Bellanger, L., Barc, J., Chatel, S., Martins, R., Mabo, P., Sacher, F., . . . Redon, R. (2015). Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet, 24(10), 2757-2763. https://doi.org/10.1093/hmg/ddv036
- Lipshultz, S., E., Law Yuk, M., Asante-Korang, A., Austin Eric, D., Dipchand Anne, I., Everitt Melanie, D., Hsu Daphne, T., Lin Kimberly, Y
- Steven, D., & null, n. (2019). Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation, 140(1), e9-e68. https://doi.org/10.1161/CIR.0000000000000682
- Lipshultz Steven, E., Law Yuk, M., Asante-Korang, A., Austin Eric, D., Dipchand Anne, I., Everitt Melanie, D., Hsu Daphne, T., Lin Kimberly, Y., Price Jack, F., Wilkinson James, D., Colan Steven, D., & null, n. (2019). Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation, 140(1), e9-e68. https://doi.org/10.1161/CIR.0000000000000682
- Magi, S., Lariccia, V., Maiolino, M., Amoroso, S., & Gratteri, S. (2017). Sudden cardiac death: focus on the genetics of channelopathies and cardiomyopathies. J Biomed Sci, 24. https://doi.org/10.1186/s12929-017-0364-6
- Maron, B. J. (2003). Sudden death in young athletes. N Engl J Med, 349(11), 1064-1075. https://doi.org/10.1056/NEJMra022783
- Maron, B. J., Gardin, J. M., Flack, J. M., Gidding, S. S., Kurosaki, T. T., & Bild, D. E. (1995). Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation, 92(4), 785-789. https://doi.org/10.1161/01.CIR.92.4.785
- Maron, B. J., Maron, M. S., & Semsarian, C. (2012). Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol, 60(8), 705-715. https://doi.org/10.1016/j.jacc.2012.02.068
- Maron, M. S. (2022, October 4). Hypertrophic cardiomyopathy: Clinical manifestations, diagnosis, and evaluation. https://www.uptodate.com/contents/hypertrophic-cardiomyopathy-clinical-manifestations-diagnosis-and-evaluation
- Martin, C. A., Huang, C. L., & Matthews, G. D. (2013). The role of ion channelopathies in sudden cardiac death: implications for clinical practice. Ann Med, 45(4), 364-374. https://doi.org/10.3109/07853890.2013.783994
- McKenna, W. J. (2024, October 8). Arrhythmogenic right ventricular cardiomyopathy: Pathogenesis and genetics. https://www.uptodate.com/contents/arrhythmogenic-right-ventricular-cardiomyopathy-pathogenesis-and-genetics
- Miller, D. T., Lee, K., Abul-Husn, N. S., Amendola, L. M., Brothers, K., Chung, W. K., Gollob, M. H., Gordon, A. S., Harrison, S. M., Hershberger, R. E., Klein, T. E., Richards, C. S., Stewart, D. R., & Martin, C. L. (2023). ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine, 25(8). https://doi.org/10.1016/j.gim.2023.100866
- Miller, D. T., Lee, K., Chung, W. K., Gordon, A. S., Herman, G. E., Klein, T. E., Stewart, D. R., Amendola, L. M., Adelman, K., Bale, S. J., Gollob, M. H., Harrison, S. M., Hershberger, R. E., McKelvey, K., Richards, C. S., Vlangos, C. N., Watson, M. S., & Martin, C. L. (2021). Correction to: ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 23(8), 1582-1584. https://doi.org/10.1038/s41436-021-01278-8
- Munroe, P. B., Addison, S., Abrams, D. J., Sebire, N. J., Cartwright, J., Donaldson, I., Cohen, M. M., Mein, C., Tinker, A., Harmer, S. C., Aziz, Q., Terry, A., Struebig, M., Warren, H. R., Vadgama, B., Fowler, D. J., Peebles, D., Taylor, A. M., Lally, P. J., & Thayyil, S. (2018). Postmortem Genetic Testing for Cardiac Ion Channelopathies in Stillbirths. https://doi.org/10.1161/CIRCGEN.117.001817
- Musunuru, K., Hershberger, R. E., Day, S. M., Klinedinst, N. J., Landstrom, A. P., Parikh, V. N., Prakash, S., Semsarian, C., & Sturm, A. C. (2020). Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circulation: Genomic and Precision Medicine, 13(4), e000067. https://doi.org/doi:10.1161/HCG.0000000000000067
- Nerbonne, J. M., & Kass, R. S. (2005). Molecular physiology of cardiac repolarization. Physiol Rev, 85(4), 1205-1253. https://doi.org/10.1152/physrev.00002.2005
- Ommen, S. R., Mital, S., Burke, M. A., Day, S. M., Deswal, A., Elliott, P., Evanovich, L. L., Hung, J., Joglar, J. A., Kantor, P., Kimmelstiel, C., Kittleson, M., Link, M. S., Maron, M. S., Martinez, M. W., Miyake, C. Y., Schaff, H. V., Semsarian, C., & Sorajja, P. (2020). 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation, 142(25), e558-e631. https://doi.org/10.1161/cir.0000000000000937
- Parker, L. E., & Landstrom, A. P. (2021). The clinical utility of pediatric cardiomyopathy genetic testing: From diagnosis to a precision medicine-based approach to care. Prog Pediatr Cardiol, 62. https://doi.org/10.1016/j.ppedcard.2021.101413
- Patton, K. K., Ellinor, P. T., Ezekowitz, M., Kowey, P., Lubitz, S. A., Perez, M., Piccini, J., Turakhia, M., Wang, P., & Viskin, S. (2016). Electrocardiographic Early Repolarization: A Scientific Statement From the American Heart Association. Circulation, 133(15), 1520-1529. https://doi.org/10.1161/cir.0000000000000388
- Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., Blom, N., Brugada, J., Chiang, C. E., Huikuri, H., Kannankeril, P., Krahn, A., Leenhardt, A., Moss, A., Schwartz, P. J., Shimizu, W., Tomaselli, G., & Tracy, C. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm, 10(12), 1932-1963. https://doi.org/10.1016/j.hrthm.2013.05.014
- Proost, D., Saenen, J., Vandeweyer, G., Rotthier, A., Alaerts, M., Van Craenenbroeck, E. M., Van Crombruggen, J., Mortier, G., Wuyts, W., Vrints, C., Del Favero, J., Loeys, B., & Van Laer, L. (2017). Targeted Next-Generation Sequencing of 51 Genes Involved in Primary Electrical Disease. J Mol Diagn, 19(3), 445-459. https://doi.org/10.1016/j.jmoldx.2017.01.010
- Roden, D. M., Balser, J. R., George, A. L., Jr., & Anderson, M. E. (2002). Cardiac ion channels. Annu Rev Physiol, 64, 431-475. https://doi.org/10.1146/annurev.physiol.64.083101.145105
- Schwartz, P. J., & Crotti, L. (2011). QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation, 124(20), 2181-2184. https://doi.org/10.1161/circulationaha.111.062182
- Schwartz, P. J., Stramba-Badiale, M., Crotti, L., Pedrazzini, M., Besana, A., Bosi, G., Gabbarini, F., Goulene, K., Insolia, R., Mannarino, S., Mosca, F., Nespoli, L., Rimini, A., Rosati, E., Salice, P., & Spazzolini, C. (2009). Prevalence of the congenital long-QT syndrome. Circulation, 120(18), 1761-1767. https://doi.org/10.1161/circulationaha.109.863209
- Seidelmann, S. B., Smith, E., Subrahmanyan, L., Dykas, D., Abou Ziki, M. D., Azari, B., Hannah-Shmouni, F., Jiang, Y., Akar, J. G., Marieb, M., Jacoby, D., Bale, A. E., Lifton, R. P., & Mani, A. (2017). Application of Whole Exome Sequencing in the Clinical Diagnosis and Management of Inherited Cardiovascular Diseases in Adults. Circ Cardiovasc Genet, 10(1). https://doi.org/10.1161/circgenetics.116.001573
- Semsarian, C., Ingles, J., Maron, M. S., & Maron, B. J. (2015). New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol, 65(12), 1249-1254. https://doi.org/10.1016/j.jacc.2015.01.019
- Spoonamore, K. G., & Johnson, N. M. (2016). Who Pays? Coverage Challenges for Cardiovascular Genetic Testing in U.S. Patients. Front Cardiovasc Med, 3. https://doi.org/10.3389/fcvm.2016.00014
- Tester, D. J., & Ackerman, M. J. (2011). Genetic Testing for Potentially Lethal, Highly Treatable Inherited Cardiomyopathies/Channelopathies in Clinical Practice. Circulation, 123(9), 1021-1037. https://doi.org/10.1161/circulationaha.109.914838
- Tester, D. J., Medeiros-Domingo, A., Will, M. L., Haglund, C. M., & Ackerman, M. J. (2012). Cardiac Channel Molecular Autopsy: Insights From 173 Consecutive Cases of Autopsy-Negative Sudden Unexplained Death Referred for Postmortem Genetic Testing. In Mayo Clin Proc (Vol. 87, pp. 524-539). https://doi.org/10.1016/j.mayocp.2012.02.017
- Towbin, J. A., McKenna, W. J., Abrams, D. J., Ackerman, M. J., Calkins, H., Darrieux, F. C. C., Daubert, J. P., de Chillou, C., DePasquale, E. C., Desai, M. Y., Estes, N. A. M., 3rd, Hua, W., Indik, J. H., Ingles, J., James, C. A., John, R. M., Judge, D. P., Keegan, R., Krahn, A. D., . . . Zareba, W. (2019). 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm, 16(11), e301-e372. https://doi.org/10.1016/j.hrthm.2019.05.007
- van Lint, F. H. M., Mook, O. R. F., Alders, M., Bikker, H., Lekanne Dit Deprez, R. H., & Christiaans, I. (2019). Large next-generation sequencing gene panels in genetic heart disease: yield of pathogenic variants and variants of unknown significance. Neth Heart J, 27(6), 304-309. https://doi.org/10.1007/s12471-019-1250-5
- Voorhees, A. P., & Han, H. C. (2015). Biomechanics of Cardiac Function. Compr Physiol, 5(4), 1623-1644. https://doi.org/10.1002/cphy.c140070
- Walsh, R., Buchan, R., Wilk, A., John, S., Felkin, L. E., Thomson, K. L., Chiaw, T. H., Loong, C. C. W., Pua, C. J., Raphael, C., Prasad, S., Barton, P. J., Funke, B., Watkins, H., Ware, J. S., & Cook, S. A. (2017). Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J, 38(46), 3461-3468. https://doi.org/10.1093/eurheartj/ehw603
- Wang, C., Hata, Y., Hirono, K., Takasaki, A., Ozawa, S. W., Nakaoka, H., Saito, K., Miyao, N., Okabe, M., Ibuki, K., Nishida, N., Origasa, H., Yu, X., Bowles, N. E., & Ichida, F. (2017). A Wide and Specific Spectrum of Genetic Variants and Genotype-Phenotype Correlations Revealed by Next-Generation Sequencing in Patients with Left Ventricular Noncompaction. J Am Heart Assoc, 6(9). https://doi.org/10.1161/jaha.117.006210
- Ware, J. S., John, S., Roberts, A. M., Buchan, R., Gong, S., Peters, N. S., Robinson, D. O., Lucassen, A., Behr, E. R., & Cook, S. A. (2013). Next generation diagnostics in inherited arrhythmia syndromes : a comparison of two approaches. J Cardiovasc Transl Res, 6(1), 94-103. https://doi.org/10.1007/s12265-012-9401-8
- Wilde, A. A. M., Veldkamp, M. W., & Smits, J. P. P. (2005). Mechanisms of inherited cardiac conduction disease. EP Europace, 7(2), 122-137. https://doi.org/10.1016/j.eupc.2004.11.004
- Yancy, C. W., Jessup, M., Bozkurt, B., Butler, J., Casey, D. E., Jr., Drazner, M. H., Fonarow, G. C., Geraci, S. A., Horwich, T., Januzzi, J. L., Johnson, M. R., Kasper, E. K., Levy, W. C., Masoudi, F. A., McBride, P. E., McMurray, J. J., Mitchell, J. E., Peterson, P. N., Riegel, B., . . . Wilkoff, B. L. (2013). 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol, 62(16), e147-239. https://doi.org/10.1016/j.jacc.2013.05.019
Coding Section
| Code |
Number |
Description |
|
| CPT |
81401 |
Molecular pathology procedure, Level 2 (e.g., 2 – 10 SNPs, 1 methylated variant, or 1 somatic variant (typically using non-sequencing target variant analysis), or detection of a dynamic mutation disorder/triplet repeat) |
|
|
|
81403 |
Molecular pathology procedure, Level 4 (e.g., analysis of single exon by DNA sequence analysis, analysis of > 10 amplicons using multiplex PCR in 2 or more independent reactions, mutation scanning or duplication/deletion variants of 2 – 5 exons) |
|
|
|
81404 |
Molecular pathology procedure, Level 7 (e.g., analysis of 11 – 25 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 26 – 50 exons, cytogenomic array analysis for neoplasia) |
|
|
|
81405 |
Molecular pathology procedure, Level 6 (e.g., analysis of 6 – 10 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 11 – 25 exons, regionally targeted cytogenomic array analysis) |
|
|
|
81406 |
Molecular pathology procedure, Level 7 (e.g., analysis of 11 – 25 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 26 – 50 exons, cytogenomic array analysis for neoplasia |
|
|
|
81407 |
Molecular pathology procedure, Level 8 (e.g., analysis of 26 – 50 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of > 50 exons, sequence analysis of multiple genes on one platform |
|
|
|
81408 |
Molecular pathology procedure, Level 9 (e.g., analysis of > 50 exons in a single gene by DNA sequence analysis) |
|
|
|
81413 |
Cardiac ion channelopathies (e.g., Brugada syndrome, long QT syndrome, short QT syndrome, catecholaminergic polymorphic ventricular tachycardia); genomic sequence analysis panel, must include sequencing of at least 10 genes, including ANK2, CASQ2, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, KCNQ1, RYR2, and SCN5A |
|
|
|
81414 |
Cardiac ion channelopathies (e.g., Brugada syndrome, long QT syndrome, short QT syndrome, catecholaminergic polymorphic ventricular tachycardia); duplication/deletion gene analysis panel, must include analysis of at least 2 genes, including KCNH2 and KCNQ1 |
|
|
|
81439 |
Hereditary cardiomyopathy (e.g., hypertrophic cardiomyopathy, dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy), genomic sequence analysis panel, must include sequencing of at least 5 cardiomyopathy-related genes (e.g., DSG2, MYBPC3, MYH7, PKP2, TTN) |
|
|
|
81479 |
Unlisted molecular pathology procedure |
|
|
|
0237U |
Medical genetics and genetic counseling services, each 30 minutes face-to-face with patient/family |
|
|
|
S3861 |
Genetic testing, sodium channel, voltage-gated, type V, alpha subunit (SCN5A) and variants for suspected brugada syndrome |
|
|
|
S3865 |
Comprehensive gene sequence analysis for hypertrophic cardiomyopathy |
|
|
|
S3866 |
Genetic analysis for a specific gene mutation for hypertrophic cardiomyopathy (HCM) in an individual with a known HCM mutation in the family |
|
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 01/27/2025 | Annual review. Updating entire policy for clarity. |
| 10/10/2024 | Moving annual review to January 2025. |
| 07/29/2024 | Change review date to 10/01/2024. |
| 04/25/2024 |
Moving review date to 7/2024. Review will coinside with Avalon's review date. |
| 04/03/2023 | Annual review no change to policy intent, but policy is being rewritten for clarity and consistency. Also updating rational and references. |
| 10/27/2022 | Annual review, no change to policy intent, but, policy rewritten for clarity and specificity. Updating rationale and references. |
| 04/07/2022 |
Annual review, no change to policy intent. Policy verbiage updated to have Short QT and Long QT syndrome written out rather than abbreviated for clarity. Also updating rationale and references. Adding table of terminology. |
| 04/15/2021 |
Annual review, updating policy to address cascade screening of first degree relatives. Also updating description, rationale and references. |
| 11/04/2020 |
Post dated on 1/9/2017 was posted in error. Disregard post dated 1/9/2017. Code 81539 was added to CAM 20433 which is Archived now. |
| 07/15/2020 |
Annual review, no change to policy intent. Updating coding. |
| 07/18/2019 |
Annual review, major revision of policy to now include information from CAM 204114 & 20228. Those policies will be archived upon the revised version of this policy being published. Updating entire policy. |
| 06/19/2019 |
Interim review. Genetic counseling is recommended is replacing Genetic counseling is Medically necessary. No other changes made. |
| 07/24/2018 |
Annual review, reformatting policy. Adding language regarding genetic counseling and adding J wave testing as investigational. |
| 07/19/2017 |
Annual review, no change to policy intent. Updating background, description, rationale and references. |
| 04/20/2017 |
Annual review, no change to policy intent. Updating background, description, rationale and references. |
| 1/9/2017 |
Updating coding with new code 81539. (Disregard this post. Code Added in error on this policy. Code 81539 should have been added to CAM 20433.) |
| 11/08/2016 |
Updating Coding section with 2017 new codes . |
| 04/27/2016 |
Annual review, updating policy to contain medical necessity criteria for Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. Updating background, description, guidelines, rationale and references. |
| 04/22/2015 |
Annual review, policy verbiage updated to include: Genetic testing for LQTS or CPVT is INVESTIGATIONAL for all other situations when the above criteria are not met. Updated background, description, guidelines, rationale and references. Added regulatory status and coding. |
| 04/03/2014 |
Annual review. Updated background, description, rationale and references. Added guidelines. Policy title being changed to Genetic Testing for Cardiac Ion Channelopathies. Added the following verbiage in the policy language: Genetic testing for CPVT may be considered medically necessary for patients who do not meet the clinical criteria for CPVT but who have:
Genetic testing for Brugada syndrome is considered INVESTIGATIONAL. Genetic testing for short QT syndrome is considered INVESTIGATIONAL. |