
Lynch Syndrome Testing - CAM 169
Description
Lynch syndrome (LS) (also known as hereditary non-polyposis colorectal cancer; HNPCC) is the most common form of hereditary colorectal (CRC) and endometrial cancers (EMC), resulting from an autosomal dominant inactivation of any of four mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) leading to microsatellite instability (MSI) and associated with an increased risk of colorectal, endometrial, stomach, small bowel, and ovarian cancers.1
For guidance concerning Tumor Mutational Burden Testing (TMB) and/or Microsatellite instability (MSI) analysis please refer to CAM 342-Microsatellite Instability and Tumor Mutational Burden Testing policy.
Regulatory Status
On Oct. 27, 2017 the FDA approved VENTANA MMR IHC Panel for patients diagnosed with colorectal cancer (CRC) to detect mismatch repair (MMR) proteins deficiency as an aid in the identification of probable Lynch syndrome and to detect BRAFV600E protein as an aid to differentiate between sporadic CRC and probable Lynch syndrome.
Additionally, many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
Consideration of both maternal and paternal family histories is necessary in the evaluation of individuals for risk of carrying a Lynch syndrome gene mutation; each lineage must be considered separately.
- For asymptomatic individuals in a family with a pathogenic familial Lynch Syndrome (LS) gene mutation who are at least 18 years of age and who have received genetic counseling, the following testing is considered MEDICALLY NECESSARY:
- Testing restricted to the known familial mutation.
- Comprehensive genetic testing, including muti-gene panel testing, when the specific familial mutation is unknown.
- For individuals with a diagnosis of any LS-related cancer (see Note 1) who have received genetic counseling, multi-gene panel testing (see Note 2, Note 3) is MEDICALLY NECESSARY when one of the following conditions is met:
- When the individual has a personal history of a tumor with MMR deficiency, determined by PCR, NGS, or IHC.
- When the individual was diagnosed before 50 years of age.
- When the individual has at any age had one or more additional LS-related cancers.
- When the individual has at least one first- or second-degree relative who was diagnosed before 50 years of age with an LS-related cancer.
- When the individual has at least two first- or second-degree relatives who have been diagnosed at any age with LS-related cancers.
- The individual has a gene mutation associated with LS-related cancers that was detected by tumor genomic profiling in the absence of germline mutation testing.
- For individuals with a known family history (see Note 4) of LS-related cancer (see Note 1) who have received genetic counseling and are at least 18 years of age, multi-gene panel testing (see Note 2, Note 3) is considered MEDICALLY NECESSARY only if the family mutation is unknown (i.e., family member is unavailable for testing or testing results are unavailable) and one of the following conditions is met:
- The individual has at least one first-degree relative who was diagnosed before 50 years of age with an LS-related cancer.
- The individual has at least one first- or second-degree relative who has been diagnosed with an LS-related cancer and another synchronous or metachronous LS-related cancer.
- The individual has at least two first- or second-degree relatives who have been diagnosed with an LS-related cancer, with at least one of the relatives having been diagnosed by 50 years of age.
- The individual has at least three first- or second-degree relatives whove have been diagnosed with LS-related cancers, regardless of their age at diagnosis.
- The individual has at least a 5% risk of having a pathogenic MMR gene variant based on predictive models (PREMM5, MMRpro, MMRpredict).
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- For all other purposes, including, but not limited to, testing of the general population, genetic testing for susceptibility to LS-related cancer is considered NOT MEDICALLY NECESSARY.
NOTES:
Note 1: According to the NCCN, “LS-related cancers include colorectal, endometrial, gastric, ovarian, pancreatic, urothelial brain (usually glioblastoma), biliary tract, and small intestine, as well as sebaceous adenomas, sebaceous carcinomas, and keratoacanthomas as seen in Muir-Torre syndrome”.2
Note 2: When germline multigene panel testing is performed in individuals with LS-related cancer, the panel should include “at minimum the following CRC, polyposis, or endometrial cancer risk-associated genes: APC, BMPR1A, BRCA1/2, EPCAM, MUTYH, MLH1, MSH2, MSH6, PMS2, PTEN, SMAD4,TP53.2
Note 3: For two or more gene tests being run on the same platform, please refer to CAM 235 Reimbursement Policy.
Note 4: Close blood relatives include first-degree relatives (i.e., parents, full siblings, and children), second-degree relatives (i.e., grandparents, aunts, uncles, nieces, nephews, grandchildren, and half-siblings), and third-degree relatives (i.e., great-grandparents, great-aunts, great-uncles, great-grandchildren, and first cousins), all of whom are on the same side of the family.
Policy Guidelines
If the tumor of the affected individual (self or family member) is available, consider initial testing of the tumor with immunohistochemistry (IHC) and/or microsatellite instability (MSI) tests
* Germline Lynch syndrome genetic testing may include testing of the gene(s) that are indicated (based on plausible eitiologies) by the abnormal tumor test result, or instead, multi-gene testing that includes MLH1, MSH2, MSH6, PMS2, and EPCAM concurrently may be performed.
Table of Terminology
| Term |
Definition |
| ACG |
American College of Gastroenterology |
| AMP |
Association for Molecular Pathology |
| APC |
Adenomatous polyposis coli |
| ASCO |
American Society of Clinical Oncology |
| ASCP |
American Society for Clinical Pathology |
| AUC |
Area under the curve |
| BLM |
BLM RecQ Like Helicase |
| BMPR1A |
Bone Morphogenetic Protein Receptor Type 1A |
| BRAC1/2 |
Breast Cancer 1/2 |
| CAP |
College of American Pathologists |
| CDN-LS |
Canadian Lynch Syndrome Working Group |
| CGA-IGC |
Collaborative Group of the Americas on Inherited Gastrointestinal Cancer |
| CHEK2 |
Checkpoint Kinase 2 |
| CLIA |
Clinical Laboratory Improvement Amendments of 1988 |
| CMS |
Centers for Medicare and Medicaid Services |
| CRC |
Colorectal cancer |
| DFCI |
Dana-Farber Cancer Institute (Harvard) |
| EGAPP |
Evaluation of Genomic Applications in Practice and Prevention |
| EMC |
Endometrial cancers |
| EPCAM |
Epithelial cellular adhesion molecule |
| ESMO |
European Society for Medical Oncology |
| FOCAD |
Focadhesin |
| GCU |
Genetic counselling unit |
| GEMCAD |
Grupo Español Multidisciplinar de Cáncer Digestivo |
| GREM1 |
Gremlin 1 |
| HNPCC |
Hereditary non-polyposis colorectal cancer |
| ICG-HNPCC |
The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer |
| IHC |
Immunohistochemistry |
| LDT |
Laboratory-developed test |
| LS |
Lynch syndrome |
| MBD4 |
Methyl-CpG Binding Domain Protein 4 |
| MGPT |
Multigene panel test |
| MLH1 |
MutL homolog 1 |
| MLH6 |
MutL homolog 6 |
| MMR |
Mismatch repair |
| MMR-D |
Mismach repair protein deficiency |
| MSH2 |
MutS homolog 2 |
| MSH6 |
MutS homolog 6 |
| MSI |
Microsatellite instability |
| MUTYH |
MutY DNA glycosylase |
| NCCN |
National Comprehensive Cancer Network |
| NCI |
National Cancer Institute |
| NGS |
Next-generation sequencing |
| NICE |
National Institute for Health and Care Excellence |
| NSGC |
National Society of Genetic Counsellors |
| NTHL1 |
Nth like deoxyribonucleic acid glycosylase 1 |
| O/E |
Observed-to-expected ratio |
| PCR |
Polymerase chain reaction |
| PMS2 |
Post meiotic segregation increased 2 (S. cerevisiae)/PMS1 homolog 2 |
| POLD1 |
Deoxyribonucleic acid polymerase delta 1 |
| POLE |
Deoxyribonucleic acid polymerase epsilon, catalytic subunit |
| PREMM5 |
Prediction model for gene mutations 5 |
| PTEN |
Phosphatase and Tensin Homolog |
| RNF43 |
Ring Finger Protein 43 |
| RPS20 |
Ribosomal Protein S20 |
| SEOM |
Spanish Society of Medical Oncology |
| SMAD |
SMAD Family Member 4 |
| STK11 |
Serine/Threonine Kinase 11 |
| TP53 |
Tumor protein 53 |
| TTD |
Grupo Español de Tumores Digestivos |
| UGI |
Upper gastrointestinal |
| USMSTF |
United States Multi-Society Task Force |
Rationale
Lynch syndrome (LS) is recognized by a hereditary predisposition to colorectal, endometrial, and other cancers due to inactivation by germline mutations or epigenetic silencing in any of four DNA mismatch repair genes—MLH1, MSH2, MSH6, and PMS2. Mutations in MLH1 and MSH2 are most common (90%) followed by MSH6 (10%) and PMS2 (6%).3 Mutations of the upstream EPCAM gene which result in silencing of the MSH2 gene produce a phenotype very similar to LS.4 LS accounts for approximately 3% to 5% of all colorectal cancers and 2% to 5% of endometrial cancers.5,6 In addition to colorectal and endometrial cancers, patients may present with ovarian, urinary tract, stomach, small bowel, hepatobiliary, sebaceous gland and central nervous system neoplasms.7
The lifetime risk of colorectal cancer (CRC) is greatly increased in LS patients but varies significantly from 10-74% dependent on which MMR gene is inactivated.8 The average age at CRC diagnosis in LS patients is 44 to 61 years with tumors primarily arising proximal to the splenic flexure.9 There is also a high rate of metachronous CRC (16% at 10 years; 41% at 20 years) in LS patients.10 The histopathology of LS colorectal cancer is often poorly differentiated with signet cell histology, abundant extracellular mucin, tumor infiltrating lymphocytes, and a lymphoid host response to tumo.11 LS patients have improved survival rates compared to similar stage spontaneous CRC (Brosens et al., 2015). .8 Lifetime risk of endometrial cancer is significantly increased to 15 – 71% of individuals with mutation specific variability.9 Increased lifetime risks have also been observed in urinary, ovarian, stomach, hepatobiliary, small bowel, brain, pancreatic and prostate cancers.8
Cancer Risks in Individuals with Lynch Syndrome Age ≤70 Years Compared to the General Population.8
| Cancer Type |
General Population Risk |
Lynch Syndrome (MLH1 and MSH2 heterozygotes) |
|
| Risk |
Mean Age of Onset |
||
| Colon |
4.8% |
52%-82% |
44-61 years |
| Endometrium |
2.7% |
25%-60% |
48-62 years |
| Stomach |
<1% |
6%-13% |
56 years |
| Ovary |
1.4% |
4%-12% |
42.5 years |
| Hepatobiliary tract |
<1% |
1.4%-4% |
Not reported |
| Urinary tract |
<1% |
1%-4% |
~55 years |
| Small bowel |
<1% |
3%-6% |
49 years |
| Brain/central nervous system |
<1% |
1%-3% |
~50 years |
| Sebaceous neoplasms |
<1% |
1%-9% |
Not reported |
Several sets of clinical criteria have been developed to identify patients with LS. In 1990, the International Collaborative Group on HNPCC established criteria (Amsterdam I Criteria) for HNPCC,12 which were updated to be more sensitive in 1999.13 The Revised Bethesda Guidelines are a third set of clinicopathologic criteria developed in 2004 to improve identification of individuals who deserve investigation for LS; however, they state, “The goal of the Bethesda Guidelines is to identify HNPCC patients, not to identify MSI-H tumors from patients in sporadic populations that may have better prognoses or different therapeutic implications”.14
Analytical Validity
Currently, there exist two main approaches to diagnosing Lynch syndrome. One approach leverages molecular screening of colorectal and endometrial tumor specimens for evidence of defective MMR function (MMR-D) or high-level MSI (MSI-H) to identify patients with cancer who should undergo germline testing for pathogenic MMR gene variants. The other focuses on using direct germline testing performed on patients whose family histories of cancer are suspicious for Lynch syndrome. In recent years, molecular testing has gained traction for identification of individuals with Lynch syndrome due to its robust sensitivity and specificity, testing of which can be generalized into one of four categories: polymerase chain reaction (PCR)-based MSI testing, immunohistochemical staining (or immunohistochemistry [IHC]) for the MMR proteins, MLH1 promoter methylation analysis (or somatic BRAF V600E mutation analysis), and next-generation somatic (and/or germline) sequencing assays.15
The specificity and sensitivity of these methods can be polemical and thus engender questions of what tests to even employ. Stinton, et al. (2021) conducted a systematic review of literature published up to August 2019 to assess the immunohistochemistry and microsatellite instability-based testing (with or without MLH1 promoter methylation testing) for Lynch syndrome in individuals with endometrial cancer. Thirteen studies consisting of approximately 3500 people were examined, and the researchers determined that, after adjusting for studies with highly selective inclusion criteria, sensitivity ranged from 60.9%-83.3% for immunohistochemistry, 69.2-89.9% for microsatellite instability-based testing, and 72.4-92.3% for studies combining immunohistochemistry, microsatellite instability-based testing, and MLH1 promoter methylation testing. According to the authors, they “found no statistically significant differences in test accuracy estimates (sensitivity, specificity) in head-to-head studies of immunohistochemistry versus microsatellite instability-based testing” and thus concluded that “sensitivity of the index tests were generally high, though most studies had much lower specificity.” However, though the authors “found no evidence that test accuracy differed between IHC and MSI based strategies,” they acknowledged that the evidence base is still quite small and at risk of bias.16
The complexity of Lynch syndrome likewise evokes the use of complex diagnostic algorithms, oftentimes involving multiple subsequent germline and somatic tests. The utility and efficacy of these algorithms are also points of contention, given the novelty of said algorithms. Through retrospectively reviewing a consecutive series of 702 patients with colorectal cancer and endometrial cancer undergoing paired tumor/germline analysis of the LS genes at a clinical diagnostic laboratory, Salvador, et al. (2019) asserted that “Paired testing identified a cause for MMRd tumors in 76% and 61% of patients without and with prior LS germline testing, respectively,” leading the researchers to support inclusion of tumor sequencing as well as comprehensive LS germline testing in the LS testing algorithm.17
Statistical models to predict risk of MMR mutations include PREMM5, MMRpredict, and MMRpro. The PREMM5 clinical prediction algorithm, “estimates the cumulative probability of an individual carrying a germline mutation in the MLH1, MSH2, MSH6, PMS2, or EPCAM genes” using an individual’s personal and family history of colorectal cancer, endometrial cancer, or other LS-related cancers with the results given as a percentage of overall predicted probability of mutation in one of the four LS-related genes .18 A study using the clinical and germline data from more than 18,000 individuals published in 2017 validated the use of the PREMM5 model. The report shows that for the four LS-related genes, PREMM5 can distinguish “carriers from noncarriers with an area under the curve (AUC) of 0.81 (95% CI, 0.79 to 0.82), and performance was similar in the validation cohort (AUC, 0.83; 95% CI, 0.75 to 0.92). Prediction was more difficult for PMS2 mutations (AUC, 0.64; 95% CI, 0.60 to 0.68) than for other genes.” The authors conclude, “ PREMM5 provides comprehensive risk estimation of all five LS genes and supports LS genetic testing for individuals with scores ≥ 2.5%”19 Kastrinos, et al. (2018) published another article the following year stating that a threshold of ≥ 2.5% is now recommended to improve the identification of PMS2 carriers by enhancing the model’s sensitivity (a threshold of ≥ 5% was previously recommended).20
MMRpro, is a statistical model and software tool that uses family history of colorectal and endometrial cancers, to estimate the likelihood of carrying a mutation in an MMR gene. It provides risk estimates that can help guide decisions about genetic testing and colorectal cancer screening.21 A study released in 2015 concluded that MMRpro was comparable to the PREMM1,2,6 model in discriminating both clinic- and population-based cohorts.22 Another study in 2017 investigated the use of MMRpro in predicting MLH1 mutations since, unlike the other LS-related genes, immunohistochemistry is less sensitive as a prescreening test for these mutations. By limiting the scope of the study to MLH1 mutations, MMRpro outperforms the PREMM1,2,6 algorithm (AUC 0.83 versus 0.68, respectively). The authors state, “Considering a threshold of 5%, MMRpro would eliminate unnecessary germline mutation analysis in a significant proportion of cases while keeping very high sensitivity. We conclude that MMRpro is useful to correctly predict who should be screened for a germline MLH1 gene mutation and propose an algorithm to improve the cost-effectiveness of LS diagnosis”.23
Likewise, the MMRpredict algorithm,is jointly operated by the Colon Cancer Genetics Group at the University of Edinburgh and MRC Human Genetics Unit of Edinburgh.24 This algorithm predicts the probability of a mutation carrier of an affected individual using criteria consisting of the age at time of diagnosis, gender, tumor location, synchronicity of tumor, and family history. A 2018 study shows that MMRpredict performs better than the PREMM5 model in identifying PMS2 mutation carriers (AUCs 0.72 and 0.51, respectively), and the efficacy of the PREMM5 model is more dependent on the location of the tumor. Both algorithms were comparable in predicting MLH1 and MSH2 mutation carriers.25 These data apparently contradict earlier findings where a previous version of the PREMM model, PREMM1,2,6, performed better than MMRpredict in predicting carriers of MLH1, MSH2, or MSH6 gene mutations. “For clinic- and population-based cohorts, O/E [observed-to-expected ratio] deviated from 1 for MMRPredict (0.38 and 0.31, respectively) and MMRpro (0.62 and 0.36) but were more satisfactory for PREMM1,2,6 (1.0 and 0.70). MMRPro or PREMM1,2,6 predictions were clinically useful at thresholds of 5% or greater and in particular at greater than 15%.”22
Mercado, et al. (2012) published a study to assess the sensitivity and specificity of PREMM1,2,6, MMRpredict, and MMRpro in 692 endometrial cancer cases (563 population-based and 129 clinic-based cases). Pathogenic mutations were identified in 3% of the population-based participants and in 62% of the clinic-based participants. “PREMM(1,2,6), MMRpredict, and MMRpro were able to distinguish mutation carriers from noncarriers (AUC of 0.77, 0.76, and 0.77, respectively), among population-based cases. All three models had lower discrimination for the clinic-based cohort, with AUCs of 0.67, 0.64, and 0.54, respectively.”26 For PREMM1,2,6, a sensitivity of 93% and a specificity of 5% was identified in population-based participants and a sensitivity of 99% and specificity of 2% was identified in clinic-based cases. For MMRpredict, a sensitivity of 71% and a specificity of 64% was identified in population-based participants and a sensitivity of 90% and specificity of 0% was identified in clinic-based cases. For MMRpro, a sensitivity of 57% and a specificity of 85% was identified in population-based participants and a sensitivity of 95% and specificity of 10% was identified in clinic-based cases.26 These authors state that the PREMM1,2,6, MMRpredict, and MMRpro seem to have limited utility in the determining which endometrial cancer patients would benefit from Lynch syndrome testing.
Clinical Utility and Validity
As use of clinical criteria and modeling to identify patients with LS has less than optimal sensitivity and can vary in efficacy between different ethnic populations,27 universal screening for LS has been recommended.28,29 Analysis by immunohistochemical testing for the MLH1/MSH2/MSH6/PMS2 proteins and/or MSI testing are commonly used to screen for LS phenotypes.30 Tumors with loss of MLH1 should undergo analysis to exclude BRAF mutation or MLH1 promoter hypermethylation according to the USPSTF.9 Moreover, patients with evidence of LS should be referred for genetic evaluation.31-33
Adar, et al. (2018) completed a study to determine the value of screening both CRC and endometrial cancer (EMC) tumors in the same population. An immunohistochemistry (IHC) screening program evaluated all patients at two centers newly diagnosed with CRC and/or EMCs. “Genetic testing was recommended for those who had tumors with absent mutS homolog 2 (MSH2), MSH6, or postmeiotoic segregation increased 2 (PMS2) expression and for those who had tumors with absent mutL homolog 1 (MLH1) expression and no v-Raf murine sarcoma viral oncogene homolog B (BRAF) mutation or MLH1 promoter methylation.”34 Scores from the PREMM1,2,6 and PREMM5 prediction models were also obtained, along with traditional Amsterdam II criteria and revised Bethesda criteria. Of the 1774 total patients screened for LS (1290 with CRC and 484 with EMC), genetic testing was recommended for 169 patients. LS was diagnosed with 16 patients with CRC and eight patients with EMC based on traditional detection methods (Amsterdam II criteria, revised Bethesda criteria, PREMM1,2,6 and PREMM5 prediction models). Of the patients genetically tested, the LS diagnosis rate was higher. Specifically, “The Amsterdam II criteria, revised Bethesda criteria, and both PREMM calculators would have missed 62.5%, 50.0%, and 12.5% of the identified patients with LS, respectively.”34 The results of this study show that risk assessment tools are likely to miss a percentage of LS diagnoses.
Laish, et al. (2021) conducted a retrospective cohort study on young patients with colorectal adenomatous polyps that aimed to “evaluate the yield of germline mutational analysis in diagnosis of LS.” All patients were 45 years or younger, with at least one adenoma removal, and underwent genetic testing by a multigene panel or LS-Jewish founder mutation panel. They found that from the 92 patients that underwent both panels, “18 patients were identified with pathogenic mutations in actionable genes, including LS-associated genes in 6 (6.5%), BRCA2 in 2 (2.5%), GREM1 in 1 (1.2%), and low-penetrance genes – APC I1307K and CHECK2- in 9 (11.4%) patients.” Generally, routine screening for establishing LS in young patients with adenomas is not recommended due to low yield, but the researchers proposed that due to these findings, genetic screening should be offered when they fulfill the clinical guidelines for LS.35
National Comprehensive Cancer Network (NCCN)
In their Genetic/Familial High-Risk Assessment: Colorectal, the NCCN lists the following criteria for the evaluation of Lynch syndrome:
- “Known LS pathogenic variant in the family
- An individual with a LS-related cancer and any of the following:
- Diagnosed <50 y
- A synchronous or metachronous LS-related cancer regardless of age
- 1 first-degree or second-degree relative with LS-related cancer diagnosed <50 y
- ≥2 first-degree or second-degree relatives with LS-related cancers regardless of age
- Family history of any of the following:
- ≥1 first-degree relative with colorectal or endometrial cancer diagnosed <50 y
- ≥1 first-degree or second-degree relative with colorectal or endometrial cancer and a synchronous or metachronous LS-related cancer regardless of age
- ≥2 first-degree or second-degree relatives with LS-related cancer including ≥1 diagnosed <50y
- ≥3 first-degree or second-degree relatives with LS-related cancers regardless of age
- Increased model-predicted risk for LS
- An individual with a ≥5% risk of having an MMR gene pathogenic variant based on predictive models (ie, PREMM5, MMRpro, MMRpredict)
- Individuals with a personal history of colorectal and/or endometrial cancer with a PREMM5 score of ≥2.5% should be considered for multi-gene panel testing.
- For individuals without a personal history of colorectal cancer and/or endometrial cancer, some data have suggested using a PREMM5 score threshold of ≥2.5% rather than ≥5% to select individuals for MMR genetic testing. Based on these data, it is reasonable for testing to be done based on the ≥2.5% score result and clinical judgment. Of note, with the lower threshold, there is an increase in sensitivity, but a decrease in specificity.
- An individual with a ≥5% risk of having an MMR gene pathogenic variant based on predictive models (ie, PREMM5, MMRpro, MMRpredict)
- Personal history of a tumor with MMR deficiency determined by PCR, NGS [next-generation sequencing], or IHC diagnosed at any age.”2
The NCCN considers LS-related cancers to “include colorectal, endometrial, gastric, ovarian, pancreas, urothelial, brain (usually glioblastoma), biliary tract, and small intestinal cancers, as well as sebaceous adenomas, sebaceous carcinomas, and keratoacanthomas as seen in Muir-Torre syndrome.” When there is no known familial mutation, the NCCN recommends germline multigene panel test (MGPT) evaluation for LS and other hereditary cancer syndromes.2
When no Lynch syndrome pathogenic variant is present in proband or in family, individuals should first refer to the Amsterdam and Bethesda criteria. However, overall, “for individuals without a previously known Lynch syndrome-associated pathogenic variant, the panel recommends additional evaluation for Lynch syndrome based on clinical criteria, including for individuals with no known Lynch syndrome pathogenic variant who meet the Amsterdam II criteria or Bethesda Guidelines, have a CRC diagnosis < 50 years of age, or have predicted risk for Lynch syndrome greater than 5% on one of the following prediction models: MMRpro, PREMM5, or MMRpredict.” When germline MGPT is performed, the panel should include “Germline MGPT should include at minimum the following CRC and/or polyposis risk-associated genes: APC, BMPR1A, EPCAM, MUTYH, MLH1, MSH2, MSH6, PMS2, PTEN, SMAD4, STK11, and TP53. Germline MGPT with the following genes that have also been associated with increased risk for polyposis and/or CRC may also be considered: monoallelic PVs in AXIN2, GREM1, POLE, and POLD1, and biallelic PVs in MSH3, MLH3, MBD4, and NTHL1… The following additional genes are found on some genetic testing panels: ATM, BLM, CHEK2, FOCAD, GALNT12, RNF43, and RPS20… Germline MGPT should include at minimum the following EC risk-associated genes: MLH1, MSH2, MSH6, PMS2, EPCAM, PTEN, and BRCA1/2.”2
In terms of initial tumor testing methodologies, “the panel recommends using only one test [either MSI or IHC testing] initially” and only “If normal results are found and Lynch syndrome is strongly suspected” that the other test be employed. Furthermore, “Where genetic testing is recommended, the panel recommends consultation with an individual with expertise in genetics, and germline testing to exclude presence of Lynch-associated P/LP variants.”2
NCCN does not recommend multi-gene testing when
- “There is an individual from a family with a known P/LP variant and there is no other reason for multi-gene testing;
- the patient’s family history is strongly suggestive of a known hereditary syndrome.”2
In these scenarios, syndrome-specific panels may be considered. For patients whose personal history is not suspicious for a polyposis syndrome and who were diagnosed with CRC ≥50 years with no known MMR deficiency in the tumor, multigene testing may be considered (category 2B). Otherwise, tumor and family history-based criteria for evaluation of Lynch syndrome is recommended for these patients.” While tumor testing can identify pathogenic/likely pathogenic variants and germline origin can sometimes be inferred with a high degree of confidence, “confirmatory germline testing is indicated for pathogenic/likely pathogenic variants with a reasonable clinical suspicion of being of germline origin (based on patient/family history or clinical characteristics, presence of founder mutation, and in some cases variant allele frequency). Somatic pathogenic/likely pathogenic variants in several genes with germline implications are common (eg, TP53, STK11, PTEN, APC), and will rarely be indicative of a need for germline testing unless clinical/family history features suggest the possibility of a germline pathogenic/likely pathogenic variant. It should be noted that the absence of reported pathogenic/likely pathogenic variants in a particular gene based on tumor testing does not rule out the possibility of a germline pathogenic/likely pathogenic variant in that gene. Clinically indicated germline testing is still appropriate for patients meeting testing guidelines regardless of tumor profiling results.”2
The NCCN states also that “In children <18 years, genetic testing is generally not recommended unless results would impact medical management, such as initiation of early colonoscopy surveillance,” though “Clear exceptions include when FAP, JPS, PJS, or constitutional MMR deficiency (CMMRD) syndrome are suspected or known to be present in a family, in which case testing prior to age 18 is recommended to guide medical management.”2
National Institute for Health and Care Excellence (NICE)
In 2017 the NICE, released their guidelines concerning molecular testing for LS in people with CRC. The recommend the following:36
- “Offer testing to all people with colorectal cancer, when first diagnosed, using immunohistochemistry for mismatch repair proteins or microsatellite instability testing to identify tumors with deficient DNA mismatch repair, and to guide further sequential testing for Lynch syndrome... Do not wait for the results before starting treatment.
- “If using immunohistochemistry, follow the steps in table 1.”
| Table 1: Steps in the immunohistochemistry testing strategy (NICE, 2017) |
||
| Step 1 |
Do an immunohistochemistry 4-panel test for MLH1, MSH2, MSH6 and PMS2. |
|
| Step 2 |
If the MLH1 immunohistochemistry result is abnormal, use sequential BRAF V600E and MLH1 promoter hypermethylation testing to differentiate sporadic and Lynch syndrome-associated colorectal cancers. First do a BRAF V600E test. |
If the MSH2, MSH6 or PMS2 immunohistochemistry results are abnormal, confirm Lynch syndrome by genetic testing of germline DNA. |
| Step 3 |
If the BRAF V600E test is negative, do an MLH1 promoter hypermethylation test. |
|
| Step 4 |
If the MLH1 promoter hypermethylation test is negative, confirm Lynch syndrome by genetic testing of germline DNA. |
|
- “If using microsatellite instability testing, follow the steps in table 2.”
| Table 2: Steps in the microsatellite instability testing strategy (NICE, 2017) |
|
| Step 1 |
Do a microsatellite instability test. |
| Step 2 |
If the microsatellite instability test result is positive, use sequential BRAF V600E and MLH1 promoter hypermethylation testing to differentiate sporadic and Lynch syndrome-associated colorectal cancers. First do a BRAF V600E test. |
| Step 3 |
If the BRAF V600E test is negative, do an MLH1 promoter hypermethylation test. |
| Step 4 |
If the MLH1 promoter hypermethylation test is negative, confirm Lynch syndrome by genetic testing of germline DNA. |
- “Healthcare professionals should ensure that people are informed of the possible implications of test results for both themselves and their relatives, and ensure that relevant support and information is available. Discussion of genetic testing should be done by a healthcare professional with appropriate training.”36
The NICE published new recommendations dealing with testing strategies for Lynch syndrome in people with endometrial cancer in 2020.37 Said recommendations are provided below.
“1.1 Offer testing for Lynch syndrome to people who are diagnosed with endometrial cancer. Use immunohistochemistry (IHC) to identify tumours with mismatch repair (MMR) deficiency:
- If IHC is abnormal with loss of MLH1, or loss of both MLH1 and PMS2 protein expression, do MLH1 promoter hypermethylation testing of tumour DNA. If MLH1 promoter hypermethylation is not detected, offer germline genetic testing to confirm Lynch syndrome.
- If IHC is abnormal with loss of MSH2, MSH6 or isolated PMS2 protein expression, offer germline genetic testing to confirm Lynch syndrome.
1.2 Healthcare professionals should inform people about the possible implications of test results for both themselves and their relatives, and give support and information. Discussion of genetic testing and obtaining consent should be done by a healthcare professional with appropriate training.
1.3 Laboratories doing IHC for MMR proteins, MLH1 promoter hypermethylation testing or germline genetic testing should take part in a recognised external quality assurance programme.”37
In February 2022, NICE updated one of their quality statements to recommend that adults with a new diagnosis of colorectal cancer be tested for Lynch syndrome. NICE recommends offering of cascade testing to family members in addition to testing individuals newly diagnosed with colorectal cancer. NICE suggests the following laboratory testing: “IHC for mismatch repair proteins or microsatellite instability testing, BRAF V600E testing and MLH1 promoter hypermethylation testing…” According to their statement, these tests should be a part of the “standard pathology report requested by oncology,” and lab providers should “ensure that laboratory protocols are in place to provide genetic testing of germline DNA for Lynch syndrome in adults with a new diagnosis of colorectal cancer and in whom test results are suggestive of Lynch syndrome.”38
American Society of Clinical Oncology (ASCO)
The ASCO recommends that “genetic testing only be conducted in the setting of pre- and post-test counseling.”39 In 2015, ASCO stated that “identifying inherited mutations in genes such as BRCA1, BRCA2, and the genes associated with Lynch syndrome allows for interventions that can significantly reduce the development of cancer and improve survival. Targeted capture assays employing NGS technology allow for testing many genes simultaneously, including genes that would not necessarily have been tested using the phenotype-directed approach, as well as genes of less clearly established clinical utility.”32 According to ASCO, multi-gene panel testing is particularly useful in situations where there are multiple high-penetrance genes associated with a specific cancer, and “one example of such a situation is Lynch syndrome, when the results of immunohistochemical analysis are not available to direct testing.”32
In 2023, ASCO endorsed the College of American Pathologists (CAP) Guidelines on testing for mismatch repair (MMR) and microsatellite instability (MSI) for patients considered for immune checkpoint inhibitor therapy: “For cancer patients being considered for immune checkpoint inhibitor therapy, if a MMR deficiency consistent with Lynch syndrome is identified in the tumor, pathologists should communicate this finding to the treating physician. (Strong recommendation).”40
U.S. Multi-Society Task Force on Colorectal Cancer (USMSTF)
The 2014 U.S. Multi-Society Task Force guidelines on colorectal cancer are captured below.
“Testing for MMR deficiency of newly diagnosed CRC should be performed. This can be done for all CRCs, or CRC diagnosed at age 70 years or younger, and in individuals older than 70 years who have a family history concerning for LS. Analysis can be done by IHC testing for the MLH1/MSH2/MSH6/PMS2 proteins and/or testing for MSI. Tumors that demonstrate loss of MLH1 should undergo BRAF testing or analysis of MLH1 promoter hypermethylation.” Also, “Individuals who have a personal history of a tumor showing evidence of MMR deficiency (without evidence of MLH1 promoter methylation); uterine cancer diagnosed at younger than age 50 years; a known family MMR gene mutation; fulfill Amsterdam criteria or revised Bethesda Guidelines; and/or have a personal risk of ≥5% chance of LS based on prediction models should undergo genetic evaluation for LS.”9
Updated 2017 guidelines from the U.S. Multi-Society Task Force give the following guideline for colorectal cancer screening and LS:41
- “colonoscopy is recommended at 10-year intervals in average-risk persons and at 1- to 2-year intervals in those with Lynch syndrome.”
However, for specific LS-related screening techniques and recommendations, the updated 2017 article states that the Giardiello, et al. (2014) guidelines are still the most current.
In 2022, the U.S. Multi-Society Task Force published updated guidelines on colorectal cancer.42 One of the recommendations was that “average-risk CRC screening” start at age 45 on a qualified basis because of the increasing incidence and mortality from colorectal cancer. However, this task force update did not mention Lynch syndrome specifically.
American Society for Clinical Pathology (ASCP), College of American Pathologists (CAP), Association for Molecular Pathology (AMP), and American Society of Clinical Oncology (ASCO)
The ASCP, CAP, AMP, and ASCO issued guidelines in 2017 stating “BRAF p.V600 mutational analysis should be performed in deficient MMR tumors with loss of MLH1 to evaluate for Lynch syndrome risk. Presence of a BRAF mutation strongly favors a sporadic pathogenesis. The absence of BRAF mutation does not exclude risk of Lynch syndrome.” In addition, they have added the following recommendation for clinicians: “clinicians should order mismatch repair status testing in patients with colorectal cancers for the identification of patients at high risk for Lynch syndrome and/or prognostic stratification.”33
American College of Gastroenterology (ACG)
In 2015, ACG issued the following practice guidelines for the management of patients with hereditary gastrointestinal cancer syndromes. The relevant guidelines are reported below.
- “All newly diagnosed colorectal cancers should be evaluated for mismatch repair deficiency.
- Analysis may be done by immunohistochemical (IHC) testing for the MLH1/MSH2/MSH6/PMS2 proteins and/or testing for microsatellite instability; tumors that demonstrate loss of MLH1 should undergo BRAF testing or analysis for MLH1 promoter hypermethylation.
- Individuals who have a personal history of a tumor showing evidence of mismatch repair deficiency (and no demonstrated BRAF mutation or hypermethylation of MLH1), a known family mutation associated with LS, or a risk of ≥5% chance of LS based on risk prediction models should undergo genetic evaluation for LS.
- Genetic testing of patients with suspected LS should include germline mutation genetic testing for the MLH1, MSH2, MSH6, PMS2, and/or EPCAM genes or the altered gene(s) indicated by IHC testing.”30
American Society of Colon and Rectal Surgeons (ASCRS)
The American Society of Colon and Rectal Surgeons published guidelines based on 2014 U.S. Multi-Society Task Force on Colorectal Cancer. There, they recommend the following:
“Universal testing (tumor testing):
- Testing for MMR deficiency of newly diagnosed CRC should be performed
- This can be done for all CRCs or CRC diagnosed at age ≤70 y and in individuals >70 y who have a family history concerning for LS
- Analysis can be done by IHC testing for the MLH1/MSH2/MSH6/PMS2 proteins and/or testing for MSI
- Tumors that demonstrate loss of MLH1 should undergo BRAF testing or analysis of MLH1 promoter hypermethylation
- To facilitate surgical planning, tumor testing on suspected CRC should be performed on preoperative biopsy specimens, if possible
Traditional testing (germline testing):
- Individuals who have a personal history of a Lynch syndrome–related tumor showing evidence of MMR deficiency (without evidence of MLH1 promoter methylation)
- Personal history of uterine cancer diagnosed at age <50 y
- A known family MMR gene mutation
- Fulfill Amsterdam criteria or revised Bethesda Guidelines
- Have a personal risk of ≥5% chance of LS based on prediction models.”43
Spanish Society of Medical Oncology (SEOM)
The SEOM published guidelines on hereditary colorectal cancer. These guidelines include the following recommendations:
- “Different screening strategies for LS of all newly diagnosed CRC and EC can be considered including tumor tests for defective MMR function and/or high-level MSI and/or NGS tumor sequencing including BRAF.
- In case of lack of expression of MLH1 and PMS2 by immunohistochemistry, BRAFV600E mutation and/or MLH1 promoter hypermethylation should be carried out to rule out sporadic cases.
- Patients with molecular profiles compatible with LS should be referred to GCU for appropriate counseling and NGS germline genetic testing.
- In families with fulfillment of rBC or a ≥ 2.5% likelihood of LS on the PREMM5 prediction model, prevalent and/or previous CRC and/or EC should follow the same screening procedure before considering referral to GCU (evidence level B, strength 1).
- Multigene panel testing for hereditary CRC and polyposis should include the genes:
- APC, BMPR1A, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4 and STK11 (evidence level A, strength 1).
- AXIN2, BLM, GREM1, NTHL1, POLD1, POLE and TP53 (evidence level B, strength 2).
- Criteria for referral to a GCU and APC/MUTYH or multigene panel testing (evidence level B, strength 1):
- Patients with > 10 synchronous adenomatous colonic polyps histologically confirmed.
- Family history of adenomatous colonic polyps (> 10 in > 1 relative), at young age and extracolonic manifestations.
- Gastric polyps (> 100), in body and fundus, preponderantly fundic glands polyps. Proton pump inhibitor use must be excluded.
- Consider in: hepatoblastoma, desmoid tumor, cribriform-morular variant of papillary thyroid carcinoma, multifocal or bilateral congenital hypertrophy of retinal pigmented epithelium.
- Known familial mutation in at-risk relatives.”44
Spanish Society of Medical Oncology (SEOM), Multidisciplinary Spanish Group of Digestive Cancer (GEMCAD), Spanish Group of Digestive Tumors (TTD)
These guidelines were the result of the consensus of ten oncologist experts in treatment from two Spanish digestive cooperative groups (Grupo Español Multidisciplinar de Cáncer Digestivo, GEMCAD, and Grupo Español de Tumores Digestivos, TTD), SEOM, and an external review panel comprising two experts designated by SEOM. Using the Infectious Diseases Society of America–U.S. Public Health Service Grading System for Ranking Recommendations in Clinical Guidelines, the collaborators assigned levels of evidence and grades of recommendation.
The authors recommend the following upon suspicion of CRC based on suggestive symptoms or screening tests:
- “A complete colonoscopy with biopsy to confirm the diagnosis is mandatory. Virtual colonoscopy is an alternative to detect potential synchronous colorectal lesions if a full colonoscopy is not feasible [I, A].
- CT scan of the chest, abdomen, and pelvis is the best technique to assess distant metastases [IV, A].
- MRI and PET-CT may be considered in selected cases [IV, B].
- Patients with mCRC should be evaluated by a multidisciplinary team to define patient management: resectable, potentially resectable, and un-resectable disease [III, A].
- The recommended staging system is that of the 8th edition of the AJCC [I, A].”45
The value of molecular profiling and identification of specific biomarkers in metastatic colon cancer are also acknowledged as predictive and prognostic indicators of disease and response to targeted therapies. As such, they note that
- “KRAS, NRAS exons 2, 3, and 4, and BRAF V600E mutations should be tested at the time of mCRC diagnosis [I, A].
- Assessment of mismatch repair deficiency (IHC or MSI-H) is recommended to assist genetic counseling for Lynch syndrome [II, B] and for its predictive value of benefit from ICI [immune checkpoint inhibition] [I, A].
- Identification of HER 2 amplification or overexpression [III, C] and NTRK fusions are recommended in subsequent lines for access to clinical trials with targeted therapies [III, A].
- Liquid biopsy might be considered to monitor emergent mutations of resistance to targeted therapy, especially prior to re-challenge with anti-epidermal growth factor receptor (anti-EGFR) treatment, though this is not supported by our national authorities [II, B].
- Testing for DPYD deficiency is strongly recommended prior to initiating 5-fluorouracil-based chemotherapy [III, A].”45
In the updated 2025 guideline, the groups expand upon their prior molecular testing recommendations by including additional biomarkers with emerging clinical relevance and tumor-agnostic indications:
- “When single or multigene tumour testing is available and applicable, testing for KRAS G12C [I, A], and POLE mutations [III, C] as well as for genomic aberrations for which targeted therapeutics are approved in tumour-agnostic indications [NTRK fusions, RET fusions, TMB-H] is advised [III, C].”46
Collaborative Group of the Americas on Inherited Gastrointestinal Cancer (CGA-IGC)
The Collaborative Group of the Americas on Inherited Gastrointestinal Cancer published a position statement on multigene panel testing for patients with colorectal cancer and/or polyposis, in which they addressed several key questions. The questions and results relevant to this policy are summarized below.
“Question 1: Which minimal set of genes should be included on a multigene panel for evaluation of hereditary CRC or polyposis?”
The CGA-IGC notes that “All commercially available panels do not include the same genes, and some panels might fail to include genes relevant to the evaluation of hereditary CRC and/or polyposis.” However, they have defined 11 genes in which PV have been associated with hereditary or familial CRC or polyposis, and “considers this list of genes to be the minimal set of genes that should clinically be tested in all patients suspected of hereditary CRC or polyposis, and recommends this testing be conducted by multigene panel.”47 It should also be noted that the authors state that “Multigene panel testing is recommended over targeted gene testing due to overlapping clinical phenotypes, inconsistent definitions for oligopolyposis, challenges with accurately classifying polyp histology (particularly with hamartomatous polyps), and variable modes of inheritance (both dominant and recessive inheritance).”47
Their table of aforementioned list of 11 genes is pulled in below.
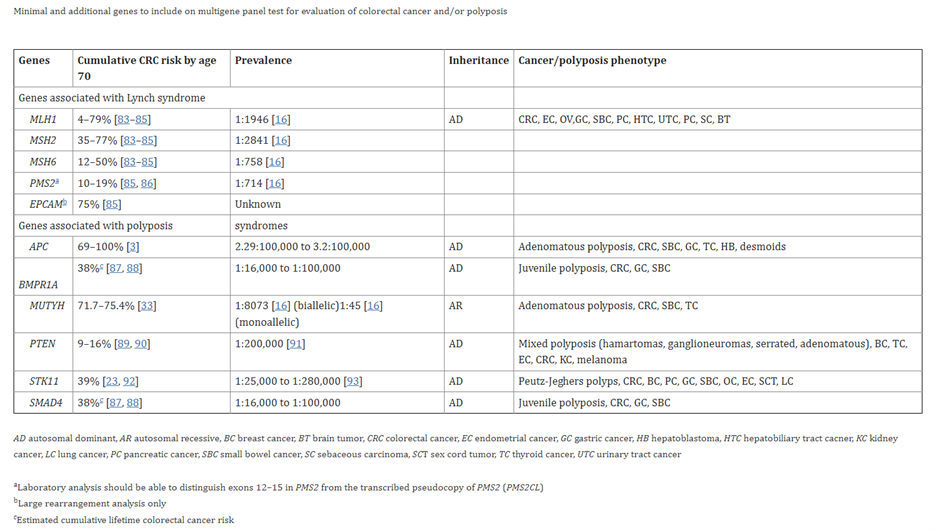
(MLH1, MSH2, MSH6, PMS2, EPCAM, APC, BMPR1A, MUTYH, PTEN, STK11, SMAD4) 47
“Question 2: Which additional set of genes should be considered on a multigene panel for evaluation of hereditary CRC or polyposis?”
According to the authors, “Beyond the minimal list of genes recommended for multigene panel testing for patients with suspected hereditary CRC, CGA-IGC recommends that 16 additional genes be considered.” Their table of 16 genes is captured below.
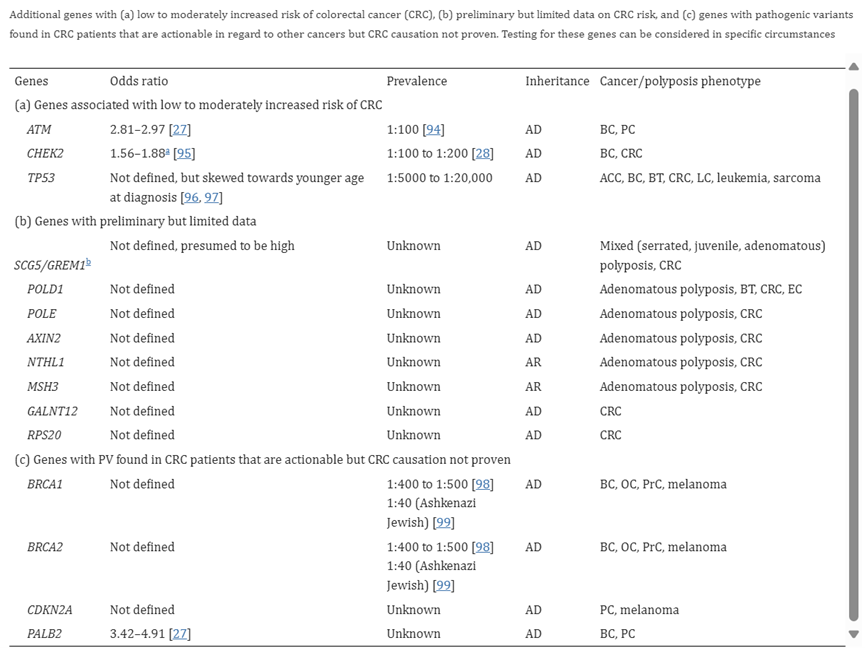
(ATM, CHEK2, TP53, POLD1, POLE, AXIN2, NTHL1, MSH3, GALNT12, BSP20, BRCA1, BRCA2, CDKN2A, PALB2)47
“Question 3: Who should undergo multigene panel testing for hereditary CRC syndromes?”
The authors state that though conventional wisdom believes that “inherited cancer syndromes only occur in those with striking personal and/or family histories of cancer,” multigene panel testing is endorsed for epithelial ovarian cancer, metastatic prostate cancer, triple negative breast cancer, and pancreatic cancer due to “the absence of typical “red flags” (family history of cancer and/or young age of onset).” Because “traditional criteria fail to identify individuals at risk for hereditary CRC,” the CGA-IGC proposes indications for panel testing in the table referenced below.

“Question 4: How should a patient with a dMMR CRC be evaluated using both germline and somatic testing?”
In the ideal situation, “the CGA-IGC endorses a simultaneous paired germline-somatic panel. However, considerations about whether to order sequential or simultaneous germline and tumor sequencing include a number of factors. If the patient’s personal and/or family history are consistent with LS, it is more likely that they will have LS, so it may be efficient to order germline testing alone given that follow-up tumor testing will probably not be necessary. If the patient has an insurance that might only cover one genetic test, it might be best to order paired tumor and normal testing since it may not be possible to reflex to tumor testing if no PV is found on germline testing. In some cases (e.g. older patients with no family history with loss of MLH1 and PMS2 without MLH1 methylation), it may actually be more likely that the patient has biallelic somatic MMR gene PV rather than a germline MMR gene PV, and paired germline and somatic testing may be a reasonable first approach to testing.” Finally, they also recommend that “patient preferences should be taken into account as some may prefer to get a complete answer quickly rather than drawing out the testing process by ordering the tests sequentially.”47
National Society of Genetic Counselors (NSGC) and the Collaborative Group of the Americas on Inherited Colorectal Cancer (CGA-IGC)
The following guidelines were provided by the NSGC and the Collaborative Group of the Americas on Inherited Colorectal Cancer:
- “Microsatellite instability (MSI) and immunohistochemistry (IHC) tumor analyses should be performed on CRC or endometrial cancers as the first‐line testing strategy for any patient being evaluated for LS (this includes individuals with CRC or endometrial cancer who meet Amsterdam I or II criteria or Bethesda Guidelines).
- MLH1 promoter methylation and BRAF V600E mutation testing may help to reduce the number of germline genetic tests needed when IHC reveals absence of MLH1 and PMS2. However, NSGC and the CGA‐ICC did not find enough data to recommend one test over the other or both concomitantly.
- IHC may occasionally yield atypical results. If IHC reveals absent MLH1 or MSH2 only, consider genetic testing of those genes individually. If IHC reveals loss of more than two MMR proteins, consider repeating the IHC analysis. If the results persist or if repeat testing was not performed, consider following the algorithm based on the most likely true results (i.e., if MSH2, MSH6 and MLH1 or PMS2 are all absent, follow the loss of MSH2/MSH6 pathway; if MLH1, PMS2 and MSH6 or MSH2 are all absent, follow the MLH1 and PMS2 pathway). Further, it is worth noting that there is a mononucleotide microsatellite in MSH6 that may cause loss of MSH6 with another MMR germline mutation leading to aberrant IHC staining patterns
- When MSI testing is stable, but IHC shows absence of one or more MMR proteins, clinical judgment should be used to determine whether tumor studies should be repeated or germline genetic testing should be pursued
- MSI testing should include, at a minimum, the five markers included in the NCI panel
- While we recognize that some third party payers may not cover MSI and/or IHC analyses on the tumor of a patient's family member(s) (e.g., the family member is deceased), in our expert opinion, we deem testing the family member(s)’ tumor is justified because: 1) LS is one of a few hereditary cancer syndromes that has a validated screening test to determine if germline genetic testing is warranted; 2) if an affected family member is living, it is likely that MSI and IHC will be covered by that relative's insurance; 3) a negative germline genetic test for all four MMR genes in an unaffected patient is uninformative; 4) the cost of direct germline genetic testing for each MMR gene ranges from $1000 to $1500, whereas the cost of MSI and IHC together is ~$1000; 5) if IHC is abnormal, additional tumor tests (BRAF and MLH1 promoter methylation) may help determine if germline genetic testing is necessary and if it is warranted, testing can be targeted to one or two genes limiting overall costs; and 6) normal MSI and IHC results on an affected individual would significantly lower the likelihood that LS is the explanation for the cancer in the family and germline genetic testing would most likely not be needed.
- Direct germline genetic testing (refers to both DNA sequencing and a technology that detects large rearrangements, insertions, deletions and duplications) may be considered on an affected or unaffected patient being evaluated for LS when MSI and IHC testing are not feasible.
- In the event that a tumor block is not available, a family member(s) is not willing or able to participate in testing, there are financial concerns or there is insufficient tissue to do either MSI or IHC testing, when indicated (e.g., high familial risk is present such as Amsterdam criteria), direct germline genetic testing may be considered. It should be noted, however, that negative germline testing in an affected individual who has not had MMR IHC can also be uninformative because there are some individuals with unidentifiable MMR gene mutations that would be followed as having LS based on abnormal IHC.”48
European Society for Medical Oncology (ESMO)
The ESMO published guidelines in 2015 for familial risk-colorectal cancer. The ASCO has endorsed these guidelines, with minor modifications.
The ASCO endorsement panel has “determined that the recommendations of the ESMO guideline are clear, thorough, and based on the most relevant scientific evidence.”49 The ASCO endorsed the ESMO guidelines (below) with a few minor qualifying statements (in bold):
- “Tumor testing for DNA mismatch repair (MMR) deficiency with immunohistochemistry for MMR proteins and/or MSI should be assessed in all CRC patients. As an alternate strategy, tumor testing should be carried out in individuals with CRC younger than 70 years, or those older than 70 years who fulfill any of the revised Bethesda Guidelines.
- If loss of MLH1/PMS2 protein expression is observed in the tumor, analysis of BRAF V600E mutation or analysis of methylation of the MLH1 promoter should be carried out first to rule out a sporadic case. If tumor is MMR deficient and somatic BRAF mutation is not detected or MLH1 promoter methylation is not identified, testing for germline mutations is indicated.
- If loss of any of the other proteins (MSH2, MSH6, PMS2) is observed, germline genetic testing should be carried out for the genes corresponding to the absent proteins (e.g., MSH2, MSH6, EPCAM, PMS2, or MLH1).
- Full germline genetic testing for Lynch syndrome should include DNA sequencing and large rearrangement analysis.
- Full germline genetic testing of APC should include DNA sequencing and large rearrangement analysis.
- Germline testing of MUTYH can be initiated by screening for the most common mutations (G396D, Y179C) in the white population followed by analysis of the entire gene in heterozygotes. Founder mutations among ethnic groups should be taken into account. For nonwhite individuals, full sequencing of MUTYH should be considered.”49
In 2019, the ESMO updated their clinical practice guidelines for hereditary gastrointestinal cancers, including those for Lynch syndrome. In this set of recommendations, the ESMO maintains that tumor testing with IHC for MMR proteins and/or MSI is recommended in individuals with CRC and that if loss of MLH1 is observed in the tumour, analysis of BRAF V600E mutation or analysis of the methylation of the MLH1 promoter should be carried out first to rule out a sporadic case. They also maintained that full germline genetic testing should include DNA sequencing and large rearrangement analysis, as in the previous guidelines, but also proposed that for those with Lynch syndrome.
- “Somatic MMR gene testing for patients with unexplained abnormal tumour screening is suggested [III, B]
- Clinical risk can be assessed using Amsterdam criteria II or the revised Bethesda Guidelines
- MMR IHC and/or MSI screening, with MLH1 promotor hypermethylation analysis in cases of MLH1 expression loss, is recommended for [individuals] with endometrial cancer [III, B]
- Follow-up recommendations in mutation carriers include colonoscopy every 1–2 years [III, A], and gynaecological examination (with TV US, CA 125 and endometrial biopsy) on a yearly basis from age 30 to 35 years [IV, C]. In all cases, age of onset in the youngest member of the family is to be considered and surveillance be started 5 years earlier [V, B]. High-quality colonoscopy carried out in dedicated centres is advised [IV, C]. UGI endoscopy surveillance (every 1–3 years, from age 30–35 years) may be considered in patients at high risk. Prophylactic gynaecological surgery might be an option for. . .carriers who have completed childbearing or are postmenopausal [IV, C].”50
In 2023, ESMO published guidelines for metastatic colorectal cancer diagnosis, treatment, and follow-up. The guidelines stated that “deficient mismatch repair (dMMR)/microsatellite instability (MSI) testing in mCRC can assist clinicians with genetic counselling, including for identification of Lynch syndrome, and should be done to select patients for immune checkpoint inhibition (ICI) as part of the initial molecular work-up.”51
Canadian Lynch Syndrome (CDN-LS) Working Group
The CDN-LS working group, composed of LS experts and patient partners, released its first official guideline in 2025. The guideline addresses universal mismatch repair (MMR), reflex testing, cascade tumor testing, and germline testing for LS. It includes several consensus-based recommendations, including the following: For universal reflex testing:
- “Universal MMR testing should be done on prospective invasive carcinoma of the colorectum, endometrium, ovarian (serous and non-serous epithelial histology), gastric, gastroesophageal junction, small bowel, ureter, renal pelvis, hepatobiliary and glioblastoma.
- Universal MMR tumour testing should NOT be done routinely on all newly diagnosed breast, prostate and bladder cancers (ie, common cancers with more limited evidence linking it to LS spectrum).
- If treatment decisions or therapeutics would be impacted by MMR status, universal testing should be done on less commonly associated LS cancers such as breast, prostate, adrenal cortical carcinoma, sarcoma and bladder cancer.”52
For cascade testing:
- “Cascade reflex MLH1 promoter methylation should be done on all endometrial cancers (EC) that are MLH1 protein deficient.
- Cascade reflex BRAF V600E and/or MLH1 promoter methylation should be done on all colorectal cancers (CRC) that are MLH1 protein deficient.
- In individuals with an LS cancer but no other clinical suspicion of LS (ie, no history of young age of diagnosis, multiple primaries, family history or polyposis):
- MMR tumour testing should be used as a first step to decrease suspicion of LS.
- In these low-risk patients, if the tumour result is MMR proficient (pMMR) or somatically explained MMR deficient (dMMR) by MLH1 promoter methylation or BRAF mutation, that is sufficient to rule out LS‡. Germline testing would not be required unless indicated for other clinical concerns (refer to statement D15).
- Somatic biallelic MMR testing (using paired tumour and blood) should be part of the testing algorithm for all unexplained dMMR patients (after ruling out BRAF, MLH1 methylation and MMR germline).”52
For germline testing:
- “In individuals with a dMMR tumour that is not somatically explained, germline multi-gene panel testing (MGPT) should include at minimum: MLH1, MSH2, MSH6, PMS2, EPCAM, MUTYH, POLE, POLD1. Consideration of larger comprehensive MGPT should be based on clinical judgement, comfort level of dealing with variants of unknown significance, cost and availability of testing at local laboratory.
- Constitutional MLH1 hypermethylation should be investigated in patients whose tumours show MLH1 promoter hypermethylation that presents early age of diagnosis (eg, <50 years), multiple MLH1-deficient primary tumours and/or suggestive family history.
- Efforts should be made to test the highest risk affected kin in the family. If the affected kin (or their sample) are unavailable, testing unaffected individuals may be considered based on suspicion of family history, risk models, availability of founder mutation testing and/or clinical judgement.
- A pMMR or somatically explained dMMR tumour cannot rule out all hereditary risks in individuals. Clinical judgement should be used to offer additional germline multi-gene panel testing if suspicion of a hereditary condition remains.”52
References
- Rumilla K, Schowalter KV, Lindor NM, et al. Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated Lynch syndrome cases. The Journal of molecular diagnostics : JMD. Jan 2011;13(1):93-9. doi:10.1016/j.jmoldx.2010.11.011
- NCCN. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2025. NCCN. Updated June 13, 2025. https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf
- Jansen M, Menko FH, Brosens LA, Giardiello FM, Offerhaus GJ. Establishing a clinical and molecular diagnosis for hereditary colorectal cancer syndromes: Present tense, future perfect? Gastrointestinal endoscopy. Dec 2014;80(6):1145-55. doi:10.1016/j.gie.2014.07.049
- Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nature genetics. Jan 2009;41(1):112-7. doi:10.1038/ng.283
- Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). The New England journal of medicine. May 05 2005;352(18):1851-60. doi:10.1056/NEJMoa043146
- Yilmaz A, Mirili C, Bilici M, Tekin SB. Colorectal cancer in Lynch syndrome associated with PMS2 and MSH6 mutations. Int J Colorectal Dis. Feb 2020;35(2):351-353. doi:10.1007/s00384-019-03454-4
- Barrow P, Khan M, Lalloo F, Evans DG, Hill J. Systematic review of the impact of registration and screening on colorectal cancer incidence and mortality in familial adenomatous polyposis and Lynch syndrome. The British journal of surgery. Dec 2013;100(13):1719-31. doi:10.1002/bjs.9316
- Brosens LA, Offerhaus GJA, Giardiello FM. Hereditary Colorectal Cancer: Genetics and Screening. Surg Clin North Am. Oct 2015;95(5):1067-80. doi:10.1016/j.suc.2015.05.004
- Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology. Aug 2014;147(2):502-26. doi:10.1053/j.gastro.2014.04.001
- Win AK, Parry S, Parry B, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Annals of surgical oncology. Jun 2013;20(6):1829-36. doi:10.1245/s10434-012-2858-5
- Peltomäki PT OG, Vasen HFA. Lynch syndrome. In: Bosman FT CF, Hruban RH, Theise ND, ed. WHO Classification of Tumours of the Digestive System. 4th ed. IARC Press; 2010.
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Diseases of the colon and rectum. May 1991;34(5):424-5. doi:10.1007/BF02053699
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999:1453-6. vol. 6.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute. Feb 18 2004;96(4):261-8. doi:10.1093/jnci/djh034
- Yurgelun MB, Hampel H. Recent Advances in Lynch Syndrome: Diagnosis, Treatment, and Cancer Prevention. American Society of Clinical Oncology Educational Book. 2018;(38):101-109. doi:10.1200/edbk_208341
- Stinton C, Fraser H, Al-Khudairy L, et al. Testing for lynch syndrome in people with endometrial cancer using immunohistochemistry and microsatellite instability-based testing strategies - A systematic review of test accuracy. Gynecol Oncol. Jan 2021;160(1):148-160. doi:10.1016/j.ygyno.2020.10.003
- Salvador MU, Truelson MRF, Mason C, et al. Comprehensive Paired Tumor/Germline Testing for Lynch Syndrome: Bringing Resolution to the Diagnostic Process. J Clin Oncol. Mar 10 2019;37(8):647-657. doi:10.1200/jco.18.00696
- DFCI. PREMM5 Model: Lynch Syndrome. Dana-Farber Cancer Institute. http://premm.dfci.harvard.edu/
- Kastrinos F, Uno H, Ukaegbu C, et al. Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. Journal of Clinical Oncology. 2017/07/01 2017;35(19):2165-2172. doi:10.1200/JCO.2016.69.6120
- Kastrinos F, Uno H, Syngal S. Commentary: PREMM5 threshold of 2.5% is recommended to improve identification of PMS2 carriers. Fam Cancer. Oct 2018;17(4):567. doi:10.1007/s10689-018-0074-6
- Harvard. MMRpro. The President and Fellows of Harvard College. 2015. https://projects.iq.harvard.edu/bayesmendel/mmrpro
- Kastrinos F, Ojha RP, Leenen C, et al. Comparison of Prediction Models for Lynch Syndrome Among Individuals With Colorectal Cancer. Journal of the National Cancer Institute. Feb 2016;108(2)doi:10.1093/jnci/djv308
- Cabreira V, Pinto C, Pinheiro M, et al. Performance of Lynch syndrome predictive models in quantifying the likelihood of germline mutations in patients with abnormal MLH1 immunoexpression. Fam Cancer. Jan 2017;16(1):73-81. doi:10.1007/s10689-016-9926-0
- Colon Cancer Genetics Group University of Edinburgh, MRC Human Genetics Unit. Prediction of DNA mismatch repair gene mutation status in incident colorectal cancer cases. Updated March 12, 2025. https://webapps.igc.ed.ac.uk/world/research/hnpccpredict/
- Goverde A, Spaander MCW, Nieboer D, et al. Evaluation of current prediction models for Lynch syndrome: updating the PREMM5 model to identify PMS2 mutation carriers. Fam Cancer. Jul 2018;17(3):361-370. doi:10.1007/s10689-017-0039-1
- Mercado RC, Hampel H, Kastrinos F, et al. Performance of PREMM(1,2,6), MMRpredict, and MMRpro in detecting Lynch syndrome among endometrial cancer cases. Genet Med. Jul 2012;14(7):670-80. doi:10.1038/gim.2012.18
- Lee SY, Kim DW, Shin YK, et al. Validation of Prediction Models for Mismatch Repair Gene Mutations in Koreans. Cancer research and treatment : official journal of Korean Cancer Association. Apr 2016;48(2):668-75. doi:10.4143/crt.2014.288
- Cohen SA, Laurino M, Bowen DJ, et al. Initiation of universal tumor screening for Lynch syndrome in colorectal cancer patients as a model for the implementation of genetic information into clinical oncology practice. Cancer. Feb 01 2016;122(3):393-401. doi:10.1002/cncr.29758
- Kidambi TD, Blanco A, Myers M, Conrad P, Loranger K, Terdiman JP. Selective Versus Universal Screening for Lynch Syndrome: A Six-Year Clinical Experience. Digestive diseases and sciences. Aug 2015;60(8):2463-9. doi:10.1007/s10620-014-3234-z
- Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. The American journal of gastroenterology. Feb 2015;110(2):223-62; quiz 263. doi:10.1038/ajg.2014.435
- EGAPP. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. Jan 2009;11(1):35-41. doi:10.1097/GIM.0b013e31818fa2ff
- Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J Clin Oncol. Nov 1 2015;33(31):3660-7. doi:10.1200/jco.2015.63.0996
- Sepulveda AR, Hamilton SR, Allegra CJ, et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. The Journal of molecular diagnostics : JMD. Mar 2017;19(2):187-225. doi:10.1016/j.jmoldx.2016.11.001
- Adar T, Rodgers LH, Shannon KM, et al. Universal screening of both endometrial and colon cancers increases the detection of Lynch syndrome. Cancer. Aug 1 2018;124(15):3145-3153. doi:10.1002/cncr.31534
- Laish I, Goldberg Y, Friedman E, et al. Genetic testing for assessment of lynch syndrome in young patients with polyps. Dig Liver Dis. Dec 2021;53(12):1640-1646. doi:10.1016/j.dld.2021.05.031
- NICE. Molecular testing strategies for Lynch syndrome in people with colorectal cancer. Updated February 22, 2017. https://www.nice.org.uk/guidance/dg27/resources/molecular-testing-strategies-for-lynch-syndrome-in-people-with-colorectal-cancer-pdf-1053695294917
- NICE. Testing strategies for Lynch syndrome in people with endometrial cancer. Updated October 28, 2020. https://www.nice.org.uk/guidance/dg42/chapter/1-Recommendations
- NICE. Quality statement 1: Testing for Lynch syndrome. Updated February 1, 2022. https://www.nice.org.uk/guidance/qs20/chapter/Quality-statement-1-Testing-for-Lynch-syndrome
- Robson ME, Storm CD, Weitzel J, Wollins DS, Offit K. American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. Feb 10 2010;28(5):893-901. doi:10.1200/jco.2009.27.0660
- Vikas P, Messersmith H, Compton C, et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: ASCO Endorsement of College of American Pathologists Guideline. Journal of Clinical Oncology. 2023;41(10):1943-1948. doi:10.1200/jco.22.02462
- Rex DK, Boland CR, Dominitz JA, et al. Colorectal Cancer Screening: Recommendations for Physicians and Patients from the U.S. Multi-Society Task Force on Colorectal Cancer. The American journal of gastroenterology. Jul 2017;112(7):1016-1030. doi:10.1038/ajg.2017.174
- Patel SG, May FP, Anderson JC, et al. Updates on Age to Start and Stop Colorectal Cancer Screening: Recommendations From the U.S. Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022/01/01/ 2022;162(1):285-299. doi:10.1053/j.gastro.2021.10.007
- Herzig DO, Buie WD, Weiser MR, et al. Clinical Practice Guidelines for the Surgical Treatment of Patients With Lynch Syndrome. Diseases of the colon and rectum. Feb 2017;60(2):137-143. doi:10.1097/dcr.0000000000000785
- Guillen-Ponce C, Lastra E, Lorenzo-Lorenzo I, et al. SEOM clinical guideline on hereditary colorectal cancer (2019). Clin Transl Oncol. Jan 24 2020;doi:10.1007/s12094-019-02272-y
- Fernández Montes A, Alonso V, Aranda E, et al. SEOM-GEMCAD-TTD clinical guidelines for the systemic treatment of metastatic colorectal cancer (2022). Clin Transl Oncol. Sep 2023;25(9):2718-2731. doi:10.1007/s12094-023-03199-1
- Montes AF, Alonso V, Aguilar EA, et al. 2025 Updated version v1.0 SEOM-GEMCAD-TTD clinical guidelines for the systemic treatment of metastatic colorectal cancer (2022). Clin Transl Oncol. Apr 2025;27(4):1845-1850. doi:10.1007/s12094-025-03860-x
- Heald B, Hampel H, Church J, et al. Collaborative Group of the Americas on Inherited Gastrointestinal Cancer Position statement on multigene panel testing for patients with colorectal cancer and/or polyposis. Fam Cancer. Jul 2020;19(3):223-239. doi:10.1007/s10689-020-00170-9
- Weissman SM, Burt R, Church J, et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. J Genet Couns. Aug 2012;21(4):484-93. doi:10.1007/s10897-011-9465-7
- Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol. Jan 10 2015;33(2):209-17. doi:10.1200/jco.2014.58.1322
- Stjepanovic N, Moreira L, Carneiro F, et al. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol. Oct 1 2019;30(10):1558-1571. doi:10.1093/annonc/mdz233
- Cervantes A, Adam, R., Rosello, S., Yoshino, T., Martinelli, E. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(1)doi:10.1016/j.annonc.2022.10.003
- Aronson M, Palma L, Semotiuk K, et al. Canadian consensus for the assessment and testing of Lynch syndrome. J Med Genet. Apr 17 2025;62(5):326-334. doi:10.1136/jmg-2024-110465
Coding Section
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2017 Forward